



Первый раз на Pharmnews.kz?
Войдите, чтобы читать, писать статьи и обсуждать всё, что происходит в мире. А также, чтобы настроить ленту исключительно под себя.
ЗарегистрироватьсяВойдите, чтобы читать, писать статьи и обсуждать всё, что происходит в мире. А также, чтобы настроить ленту исключительно под себя.
Зарегистрироваться












03 ноября 2016


Решение Совета Евразийской экономической комиссии № 77 от 3 ноября 2016 года
Об утверждении Правил надлежащей производственной практики Евразийского экономического союза
В соответствии со статьями 30 и 56 Договора о Евразийском экономическом союзе от 29 мая 2014 года, пунктом 14 приложения № 12 к Договору о Евразийском экономическом союзе от 29 мая 2014 года, статьей 9 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, пунктами 57 и 82 приложения № 1 к Регламенту работы Евразийской экономической комиссии, утвержденному Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. № 98, и Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. № 108 «О реализации Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза» Совет Евразийской экономической комиссии решил:
1. Утвердить прилагаемые Правила надлежащей производственной практики Евразийского экономического союза (далее - Правила).
2. Настоящее Решение вступает в силу по истечении 10 календарных дней с даты вступления в силу Протокола, подписанного 2 декабря 2015 года, о присоединении Республики Армения к Соглашению о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, но не ранее чем по истечении 10 календарных дней с даты официального опубликования настоящего Решения, за исключением положений утвержденных настоящим Решением Правил, касающихся требований к производству ветеринарных лекарственных средств.
Положения Правил, касающиеся требований к производству ветеринарных лекарственных средств, вступают в силу с 1 января 2021 г.
Члены Совета Евразийской экономической комиссии:
|
От Республики Армения |
От Республики Беларусь |
От Республики Казахстан |
От Кыргызской Республики |
От Российской Федерации |
|
В. Габриелян |
В. Матюшевский |
А. Мамин |
О. Панкратов |
И. Шувалов |
УТВЕРЖДЕНЫ
Решением Совета
Евразийской экономической комиссии
от 3 ноября 2016 г. № 77
ПРАВИЛА
надлежащей производственной практики Евразийского экономического союза
ВВЕДЕНИЕ
Фармацевтическая промышленность государств - членов Евразийского экономического союза (далее соответственно - государства-члены, Союз) поддерживает высокие стандарты управления качеством при разработке, производстве и контроле лекарственных средств. Система государственной регистрации гарантирует, что все лекарственные средства оценены уполномоченным органом, чтобы обеспечить их соответствие современным требованиям безопасности, качества и эффективности. Система лицензирования производства гарантирует, что вся продукция, разрешенная к применению на территориях государств-членов, произведена только производителями, которые имеют соответствующие разрешения (лицензии) и регулярно инспектируются уполномоченными органами с использованием принципов управления рисками для качества. Разрешение (лицензия) на производство является обязательным для производителя лекарственных средств государств-членов независимо от того, реализуется эта продукция на территориях государств-членов или за их пределами.
Соответствие настоящим Правилам учитывается при получении разрешений (лицензий) на производство лекарственных средств, и на нем основывается инспектирование производителей лекарственных средств.
Требования настоящих Правил к производству ветеринарных лекарственных средств являются такими же, как и при производстве лекарственных средств для медицинского применения. Специальные требования настоящих Правил для ветеринарных лекарственных препаратов и иммунобиологических ветеринарных лекарственных препаратов изложены в приложениях № 4 и 5.
Настоящие Правила представлены 3 частями и рядом приложений. Часть I содержит принципы, применимые при производстве лекарственных препаратов. Часть II охватывает принципы, применимые при производстве активных фармацевтических субстанций, используемых в качестве исходных материалов. Часть III содержит разделы, в которых разъясняются требования уполномоченных органов государств-членов, связанные с правилами надлежащего производства лекарственных средств.
В главе 1 части I настоящих Правил в общих чертах излагается фундаментальная концепция управления качеством при производстве лекарственных препаратов. Каждая из следующих глав содержит принцип, описывающий в общих чертах цели управления качеством в рамках этой главы, и пояснения, которые обеспечивают достаточную детализацию, чтобы производители понимали основные вопросы, которые необходимо учитывать при реализации этого принципа.
В настоящих Правилах излагается детальная информация о принципах надлежащего производства в отношении активных фармацевтических субстанций, используемых в качестве исходных материалов. Часть II была разработана на основе руководства ICH, изданного как документ ICH Q7A для активных фармацевтических субстанций. Эта часть распространяется как на лекарственные средства для медицинского применения, так и на ветеринарные лекарственные средства.
В дополнение к основному содержанию, изложенному в частях I и II, настоящие Правила включают в себя ряд приложений, обеспечивающих детализацию в отношении отдельных видов деятельности. Для некоторых производственных процессов различные приложения будут применяться одновременно (например, приложения, регламентирующие производство стерильных лекарственных средств и производство радиофармацевтических лекарственных средств и (или) биологических лекарственных средств).
Часть III содержит разделы, связанные с правилами надлежащего производства, которые не являются подробными руководящими принципами. Цель части III - разъяснить требования регуляторных органов, ее следует рассматривать как источник информации в отношении наилучших современных методов. Отдельно в каждом разделе описаны детали, касающиеся его применимости.
После приложений к основным частям Правил приведен словарь терминов, используемых в настоящих Правилах.
Настоящие Правила не распространяются на вопросы охраны труда персонала, занятого в производстве. Эти вопросы могут быть важными при производстве таких лекарственных средств, как высокоактивные, биологические и радиоактивные. Они регулируются законодательством государств-членов.
Настоящими Правилами предусматривается, что держатель разрешения (лицензии) на производство систематически включает требования регистрационного досье лекарственного препарата в отношении безопасности, качества и эффективности продукции во все мероприятия по производству, контролю и выпуску продукции в реализацию.
В течение многих лет производство лекарственных средств проводится в соответствии с руководящими принципами правил надлежащего производства и не регулируется стандартами CEN/ISO. В настоящих Правилах стандарты CEN/ISO были учтены, но терминология этих стандартов не применялась.
Могут существовать иные приемлемые методы, отличные от описанных в настоящих Правилах, с помощью которых можно соблюсти принципы управления качеством. Настоящие Правила не направлены на ограничение развития каких-либо новых концепций или новых технологий, которые прошли валидацию и обеспечивают уровень управления качеством, по меньшей мере эквивалентный установленному настоящими Правилами.
Настоящие Правила будут регулярно пересматриваться с целью отражения непрерывного совершенствования практики в области качества.
I. Основные требования
Глава 1. Фармацевтическая система качества
Принцип
Производитель должен производить лекарственные средства таким образом, чтобы гарантировать их соответствие своему назначению, требованиям регистрационного досье и (или) протоколу клинического исследования и минимизировать риск для пациентов, связанный с безопасностью, качеством и эффективностью лекарственных средств. Ответственность за выполнение этих требований несет высшее руководство, их выполнение требует участия и ответственности персонала различных подразделений предприятия-производителя на всех его уровнях, а также поставщиков и организаций оптовой торговли. Для достижения этой цели создается всесторонне разработанная и правильно функционирующая фармацевтическая система качества, включающая в себя надлежащую производственную практику и управление рисками для качества. Эта система должна быть оформлена документально, а ее эффективность - проконтролирована. Все элементы фармацевтической системы качества должны быть укомплектованы квалифицированным персоналом, обеспечены необходимыми и надлежащими помещениями, оборудованием и техническими средствами. Держатель разрешения (лицензии) на производство лекарственных средств и уполномоченное лицо (лица) несут ответственность в соответствии с законодательством государств-членов за функционирование фармацевтической системы качества.
Основные принципы управления качеством, надлежащей производственной практики и управления рисками для качества взаимосвязаны. Они описаны ниже, чтобы подчеркнуть их взаимосвязь и первостепенное значение для производства и контроля лекарственных препаратов.
Фармацевтическая система качества
1.1. Управление качеством - всеобъемлющее понятие, охватывающее все вопросы, которые в отдельности или в целом влияют на качество продукции. Это совокупность организационных мер, предпринимаемых в целях обеспечения соответствия качества лекарственных средств их предназначению. Управление качеством включает в себя надлежащую производственную практику.
1.2. Правила надлежащего производства и контроля качества применяются ко всем стадиям жизненного цикла продуктов: производству лекарственных препаратов для клинических исследований, переносу технологии, промышленному производству, прекращению производства лекарственных средств. Однако фармацевтическая система качества может распространяться и на такую стадию жизненного цикла продуктов, как фармацевтическая разработка. Это описано в части III настоящих Правил в разделе «Фармацевтическая система качества», который, несмотря на то, что является рекомендательным, должен способствовать инновациям и постоянному улучшению, а также упрочнению связи между фармацевтической разработкой и производственной деятельностью. Раздел «Фармацевтическая система качества» может использоваться, чтобы дополнить содержание настоящей главы.
1.3. При разработке новой фармацевтической системы качества или при изменении существующей системы необходимо учитывать объем и сложность деятельности организации. В структуру фармацевтической системы качества должны быть включены соответствующие принципы управления рисками с использованием подходящих инструментов. В то время как некоторые аспекты фармацевтической системы качества могут применяться к деятельности всей организации в целом, а иные - только к определенным производственным участкам, эффективность внедрения фармацевтической системы качества обычно демонстрируется на уровне производственной площадки.
1.4. Фармацевтическая система качества, предназначенная для производства лекарственных средств, должна гарантировать, что:
i) выпуск продукции обеспечивается посредством разработки, планирования, внедрения, поддержания и непрерывного усовершенствования системы, которая дает возможность постоянно поставлять продукцию с соответствующими показателями качества;
ii) управление знаниями о продукции и процессе ее производства осуществляется на протяжении всех стадий жизненного цикла продукции;
iii) лекарственные средства разрабатываются и совершенствуются с учетом требований настоящих Правил;
iv) операции по производству и контролю точно определены и соответствуют требованиям настоящих Правил;
v) ответственность и обязанности руководителей четко определены;
vi) приняты меры для производства, поставки и использования исходных материалов, соответствующих установленным требованиям, а также для выбора и контроля поставщиков и для проверки того, что каждая поставка получена от утвержденных поставщиков (утвержденной цепи поставки);
vii) внедрены процессы, обеспечивающие управление деятельностью, передаваемой другой организации для выполнения (аутсорсинговой деятельностью);
viii) установлено и поддерживается контролируемое состояние посредством разработки и использования эффективного мониторинга и систем контроля в отношении проведения процесса и качества продукции;
ix) результаты мониторинга процессов и качества продукции принимаются во внимание при выпуске серии, при расследовании отклонений и для принятия предупреждающих мер во избежание потенциальных отклонений, которые могут произойти в будущем;
x) проводятся необходимый контроль промежуточной продукции, любой другой контроль в процессе производства, и осуществляется валидация;
xi) оказывается содействие постоянному улучшению посредством внедрения усовершенствований, основанных на актуальных знаниях процесса и продукции;
xii) приняты меры для перспективной оценки запланированных изменений и их утверждения до внедрения с учетом необходимости выполнить уведомление уполномоченных органов или согласовать изменения с ними, если это требуется;
xiii) проводится оценка любых реализованных изменений после их внедрения для подтверждения того, что цель изменений была достигнута и что это не отразилось на качестве продукции негативно;
xiv) во время расследования отклонений, предполагаемых дефектов продукции и других проблем должен применяться соответствующий уровень анализа основных причин данных несоответствий, который может быть определен с использованием принципов управления рисками для качества. В случаях, когда истинная основная причина (причины) несоответствия не может быть установлена, следует идентифицировать наиболее вероятную причину. В случаях, когда в качестве причины подозревается или идентифицирована ошибка персонала, это должно быть доказано с особой тщательностью, чтобы гарантировать, что не были пропущены существующие процессные, процедурные или системные ошибки или проблемы. По результатам расследования должны быть определены и предприняты соответствующие корректирующие и (или) предупреждающие действия. Эффективность таких действий должна быть проверена и оценена в соответствии с принципами управления рисками для качества;
xv) лекарственные препараты не будут выпущены в обращение до того, как уполномоченное лицо не удостоверит, что каждая серия продукции была произведена и проконтролирована в соответствии с требованиями регистрационного досье и настоящих Правил;
xvi) предприняты достаточные меры, обеспечивающие поддержание качества лекарственных препаратов в течение всего срока годности при их хранении и последующем обращении;
xvii) имеется процедура проведения самоинспекции и (или) аудита качества, в соответствии с которой регулярно оцениваются эффективность и пригодность фармацевтической системы качества.
1.5. Высшее руководство несет основную ответственность за наличие эффективной фармацевтической системы качества, за то, что имеются необходимые ресурсы и что обязанности, ответственность и полномочия определены, доведены до сведения и выполняются, реализуются во всех подразделениях предприятия. Важна лидирующая роль высшего руководства и его активное участие в фармацевтической системе качества. Это должно гарантировать поддержку фармацевтической системы качества и заинтересованность персонала на всех уровнях и в подразделениях предприятия.
1.6. Должны проводиться периодические обзоры функционирования фармацевтической системы качества с вовлечением в этот процесс высшего руководства, чтобы определить возможности для постоянного улучшения продукции, процессов и самой системы.
1.7. Фармацевтическая система качества должна быть определена и оформлена документально. Должно быть в наличии руководство по качеству или эквивалентный ему документ, который должен содержать описание системы управления качеством, включая ответственность руководства.
Надлежащая производственная практика
1.8. Надлежащая производственная практика является той частью управления качеством, которая гарантирует, что продукция постоянно производится и контролируется по стандартам качества, соответствующим ее назначению, а также в соответствии с требованиями регистрационного досье, протокола клинических исследований и спецификации на эту продукцию.
Надлежащая производственная практика связана как с производством, так и с контролем качества. Основные требования надлежащей производственной практики:
i) все производственные процессы определяются, систематически пересматриваются с учетом накопленного опыта и подтверждают способность постоянно производить лекарственные препараты требуемого качества в соответствии со спецификациями;
ii) критические стадии производственного процесса и существенные изменения процесса должны пройти валидацию;
iii) должны быть обеспечены все необходимые условия для выполнения требований настоящих Правил, включая наличие:
надлежащим образом обученного персонала, имеющего необходимую квалификацию;
соответствующих помещений и площадей;
соответствующих оборудования и обслуживания;
соответствующих материалов, контейнеров и этикеток;
утвержденных процедур и инструкций в соответствии с фармацевтической системой качества;
соответствующих условий хранения и транспортирования;
iv) инструкции и процедуры должны быть изложены в письменной форме ясно и недвусмысленно, они должны быть конкретно применимы к имеющимся в наличии средствам;
v) процедуры должны точно соблюдаться, и персонал должен быть обучен правильному их выполнению;
vii) в процессе производства следует составлять записи (рукописным способом и (или) с применением технических средств), документально подтверждающие фактическое проведение этапов, предусмотренных установленными методиками и инструкциями, а также то, что количество и качество продукции соответствуют установленным нормам;
viii) любые существенные отклонения должны быть полностью оформлены документально и расследованы с целью определения причины отклонения и осуществления соответствующих корректирующих и предупреждающих действий;
ix) в понятной и доступной форме сохраняются записи, относящиеся к серии (например, в досье на серию), включая документацию по реализации, которые позволяют проследить полную историю серии;
x) при оптовой реализации продукции необходимо свести к минимуму риски для ее качества и учитывать правила надлежащей дистрибьюторской практики, утверждаемые Евразийской экономической комиссией;
xi) должна быть в наличии система отзыва любой серии продукции из продажи или поставки;
xii) должны рассматриваться претензии на поставленную продукцию, расследоваться причины дефектов и приниматься соответствующие меры как в отношении недоброкачественной продукции, так и для предотвращения подобных случаев.
Контроль качества
1.9. Контроль качества является частью надлежащей производственной практики, связанной с отбором проб, спецификациями и проведением испытаний, а также с процедурами организации, документирования и выдачи разрешения на выпуск, гарантирующими, что фактически проведены все необходимые испытания и что материалы не будут разрешены для использования, а готовая продукция не будет допущена к реализации или поставке до тех пор, пока их качество не будет признано удовлетворительным.
Основные требования к контролю качества:
i) должны быть в наличии соответствующие помещения и оборудование, обученный персонал и утвержденные методики для отбора проб, контроля и испытаний исходных материалов, промежуточной, нерасфасованной и готовой продукции, а также (при необходимости) для мониторинга условий производственной среды в целях выполнения требований настоящих Правил;
ii) проведение отбора проб исходных и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции назначенным персоналом в соответствии с утвержденными методиками;
iii) методы испытаний должны пройти валидацию;
iv) должны быть составлены записи (рукописным способом и (или) с применением технических средств), документально подтверждающие, что все необходимые мероприятия по отбору проб, контролю и методикам испытаний действительно проведены. Любые отклонения должны быть полностью оформлены документально и расследованы;
v) готовая продукция должна содержать активные фармацевтические субстанции, соответствующие регистрационному досье по качественному и количественному составу, должна иметь требуемую чистоту, должна быть вложена в надлежащую упаковку и правильно маркирована;
vi) записи, оформленные по результатам контроля и испытаний материалов, промежуточной, нерасфасованной и готовой продукции, должны быть официально сопоставлены с требованиями спецификаций. Оценка продукции должна включать в себя обзор и оценку соответствующей производственной документации и оценку отклонений от установленных процедур;
vii) ни одна серия продукции не может быть разрешена для реализации или поставки до того, как уполномоченное лицо согласно приложению № 16 не удостоверит ее соответствие требованиям, установленным при государственной регистрации;
viii) необходимо сохранять достаточное количество контрольных образцов исходных и упаковочных материалов и готовой продукции, которое позволит проводить испытания в будущем (при необходимости) согласно приложению № 19. Образцы готовой продукции следует хранить в окончательной упаковке.
Обзор качества продукции
1.10. Следует регулярно проводить обзоры качества всех зарегистрированных произведенных лекарственных препаратов, в том числе лекарственных препаратов, изготовляемых только на экспорт, с целью подтверждения постоянства имеющегося процесса, соответствия действующим спецификациям как на исходные материалы, так и на готовую продукцию, чтобы выявить какие-либо тенденции (тренды) и установить возможность усовершенствования продукции и процесса. Такие обзоры следует оформлять документально и проводить, как правило, ежегодно, принимая во внимание предыдущие обзоры. Они должны включать, как минимум, следующее:
i) обзор исходных материалов (включая упаковочные материалы), используемых при производстве, особенно отмечая те, которые получены от новых поставщиков, и отдельный обзор прослеживаемости цепи поставки активных фармацевтических субстанций;
ii) обзор критических точек контроля в процессе производства и результатов контроля готовой продукции;
iii) обзор всех серий, которые не соответствовали установленным спецификациям, и результатов соответствующих расследований;
iv) обзор всех существенных отклонений или несоответствий, связанных с ними расследований, эффективности и результативности предпринятых корректирующих и предупреждающих действий;
v) обзор всех изменений, внесенных в процессы или аналитические методики;
vi) обзор поданных, утвержденных или отклоненных изменений в регистрационное досье, в том числе в досье на лекарственные препараты, предназначенные только для экспорта;
vii) обзор результатов мониторинга стабильности и любых неблагоприятных тенденций;
viii) обзор всех связанных с качеством продукции возвратов, претензий и отзывов, а также проведенных в это время расследований;
ix) обзор достаточности любых ранее проведенных корректирующих действий в отношении производства или оборудования;
x) обзор пострегистрационных обязательств при получении новых регистрационных удостоверений или внесения изменений в регистрационные досье;
xi) состояние квалификации соответствующих оборудования и технических средств, например, систем нагрева, вентиляции и кондиционирования воздуха, систем снабжения водой, сжатыми газами и т. д.;
xii) обзор любых контрактных соглашений, указанных в главе 7 настоящих Правил, с целью подтверждения их соответствия действующим требованиям.
1.11. Производитель и держатель регистрационного удостоверения (если они являются разными организациями) должны оценивать результаты обзора качества и делать выводы о необходимости осуществления корректирующих и предупреждающих действий или проведения повторной валидации в рамках фармацевтической системы качества. Должны быть в наличии процедуры для постоянного управления и анализа таких действий. Эффективность этих процедур должна быть подтверждена во время самоинспекции. Обзоры качества можно группировать по видам продукции (например, твердые лекарственные формы, жидкие лекарственные формы, стерильные лекарственные препараты и т. д.) при наличии научного обоснования.
Если держатель регистрационного удостоверения не является производителем, то должно быть заключено соглашение между ним и производителем, в котором установлены соответствующие обязанности сторон в отношении составления обзора качества.
Управление рисками для качества
1.12. Управление рисками для качества является систематизированным процессом оценки, контроля, передачи информации, а также обзора рисков для качества лекарственного препарата. Этот процесс может проводиться как перспективно, так и ретроспективно.
1.13. Принципы управления рисками для качества:
оценка рисков для качества основывается на научных знаниях, опыте работы в отношении процесса и в конечном счете связана с защитой пациента;
масштаб работ, степень формализации и документального оформления процесса управления рисками для качества соответствуют уровню риска.
Примеры процессов и применения управления рисками для качества также указаны в главе II части III настоящих Правил.
Глава 2. Персонал
Принцип
Надлежащее производство лекарственных средств зависит от персонала. Поэтому на предприятии должно быть достаточное количество квалифицированного персонала для решения всех задач, относящихся к сфере ответственности производителя. Каждый сотрудник должен понимать индивидуальную ответственность, которая должна быть документально оформлена. Весь персонал должен знать принципы надлежащей производственной практики, касающиеся его деятельности, а также пройти первичное и последующее обучение в соответствии с его обязанностями, включая инструктаж по выполнению гигиенических требований.
Общие требования
2.1. Производитель должен иметь достаточное количество сотрудников с необходимой квалификацией и практическим опытом работы. Высшее руководство должно определять и обеспечивать достаточные и надлежащие ресурсы (человеческие, финансовые, материальные, а также помещения и оборудование) для внедрения и поддержания системы управления качеством и постоянного повышения ее эффективности. Обязанности любого сотрудника не должны быть чрезмерными, чтобы исключить возможность возникновения рисков для качества продукции.
2.2. На предприятии должна иметься схема организационной структуры, в которой отношения между руководителями производства, контроля качества и, где это применимо, руководителем подразделения обеспечения качества или подразделения по качеству, указанные в пункте 2.5 настоящего раздела, и позиция уполномоченного лица (лиц) ясно обозначены в иерархии управления.
2.3. Обязанности и соответствующие полномочия для их выполнения должны быть определены и прописаны в должностных инструкциях. Обязанности руководящего персонала могут быть переданы назначенным заместителям, обладающим достаточным уровнем квалификации. Круг обязанностей персонала должен охватывать все стороны надлежащей производственной практики, однако не должно быть необоснованного дублирования сфер ответственности.
2.4. Высшее руководство несет основную ответственность за обеспечение эффективной системы управления качеством для достижения целей в области качества, а также за то, что роли, обязанности и полномочия определены, доведены до сведения персонала и осуществляются в рамках всей организации. Высшее руководство должно установить политику в области качества, которая определяет общие намерения и направления деятельности компании, связанные с качеством, и должно обеспечить постоянную пригодность и эффективность системы управления качеством и соответствие надлежащей производственной практики путем участия в анализе со стороны руководства.
Ключевой персонал
2.5. Высшее руководство должно назначить ключевой руководящий персонал, включая руководителя производства и руководителя подразделения контроля качества, а также достаточное число (но не менее 1) уполномоченных лиц, если в обязанности руководителя производства и (или) руководителя подразделения контроля качества не входят обязанности, предусмотренные для уполномоченного лица. Ключевой руководящий персонал, как правило, должен работать полный рабочий день. Руководители производства и подразделения контроля качества должны быть независимы друг от друга. В больших организациях может возникнуть необходимость передать другим сотрудникам отдельные обязанности, указанные в пунктах 2.7 - 2.9 настоящего раздела.
Кроме того, в зависимости от размера и структуры организации может быть отдельно назначен руководитель подразделения обеспечения качества или руководитель подразделения качества. При наличии таких подразделений некоторые их обязанности, указанные в пунктах 2.7 - 2.9 настоящего раздела, будут общими с обязанностями руководителя подразделения контроля качества и руководителя производства, поэтому высшее руководство должно обеспечить, чтобы их обязанности и полномочия были четко и недвусмысленно определены.
2.6. Обязанности уполномоченного лица:
для лекарственных средств, произведенных на территориях государств-членов, уполномоченное лицо должно гарантировать, что каждая серия была произведена и проверена в соответствии с законодательством этих государств и в соответствии с требованиями регистрационного досье;
для лекарственных средств из третьих стран независимо от того, были ли они произведены на территориях государств-членов, уполномоченное лицо должно гарантировать, что каждая производственная серия подверглась в государстве-члене полному качественному и количественному анализу по крайней мере всех активных веществ и всем другим испытаниям или проверкам, необходимым для обеспечения качества лекарственных средств в соответствии с требованиями регистрационного досье.
Уполномоченное лицо (лица) перед выпуском любой серии должно документально подтвердить, что все необходимые операции выполнены и что каждая серия соответствует установленным требованиям.
Образование, обучение и стаж работы уполномоченных лиц должны соответствовать требованиям, установленным международными договорами и решениями органов Союза, составляющим право Союза. Они должны постоянно и непрерывно находиться в распоряжении владельца разрешения (лицензии) на производство для выполнения своих обязанностей.
Обязанности уполномоченного лица могут быть переданы только другому уполномоченному лицу (лицам).
Порядок подтверждения уполномоченным лицом соответствия серии продукции приведен в приложении № 16.
2.7. Обязанности руководителя производства включают в себя, как правило:
i) обеспечение соответствия производства и хранения продукции утвержденной документации для обеспечения требуемого качества;
ii) утверждение инструкций, относящихся к производственным операциям, и обеспечение их строгого выполнения;
iii) обеспечение оценки и подписания производственных документов уполномоченным на это персоналом;
iv) обеспечение и гарантию квалификации, надлежащего содержания, эксплуатации и технического обслуживания помещений и оборудования в своем подразделении;
v) обеспечение и гарантию проведения соответствующей валидации;
vi) обеспечение и гарантию проведения необходимого первичного и последующего непрерывного обучения персонала своего подразделения.
2.8. Обязанности руководителя подразделения контроля качества включают в себя, как правило:
i) одобрение или отклонение, если он считает это необходимым, исходного сырья и упаковочных материалов, а также промежуточной, нерасфасованной и готовой продукции;
ii) обеспечение проведения всех необходимых испытаний и оценку соответствующих записей;
iii) утверждение спецификаций, процедур по отбору проб, методик испытаний и других процедур по контролю качества;
iv) одобрение специалистов, привлекаемых к проведению испытаний по контракту, и осуществление контроля за ними;
v) обеспечение и гарантию квалификации, надлежащего содержания, эксплуатации и технического обслуживания помещений и оборудования в своем подразделении;
vi) обеспечение и гарантию проведения соответствующей валидации;
vii) обеспечение и гарантию проведения необходимого первичного и последующего непрерывного обучения персонала своего подразделения.
Другие обязанности персонала подразделения контроля качества указаны в главе 6 части I настоящих Правил.
2.9. Руководители производства и подразделения контроля качества, а также руководитель отдела обеспечения качества или руководитель службы качества (при необходимости), как правило, имеют некоторые общие или совместно выполняемые обязанности, относящиеся к обеспечению качества продукции, включая, в частности, разработку, эффективное внедрение, поддержание и мониторинг системы управления качеством.
Эти обязанности могут включать в себя:
i) согласование и утверждение письменных процедур и других документов, в том числе внесение в них изменений;
ii) мониторинг и контроль производственной среды;
iii) контроль за соблюдением гигиенических требований на предприятии;
iv) валидацию процессов;
v) обучение персонала;
vi) утверждение и мониторинг поставщиков исходных и упаковочных материалов;
vii) утверждение и мониторинг организаций, выполняющих работы по контракту, и поставщиков других связанных с надлежащей производственной практикой аутсорсинговых услуг;
viii) определение и мониторинг соблюдения условий хранения материалов и продукции;
ix) хранение записей;
x) мониторинг соблюдения требований GMP;
xi) проверку, расследование и отбор проб (образцов) в целях контроля факторов, которые могут повлиять на качество продукции;
xii) участие в анализе со стороны руководства функционирования процессов, качества продукции и системы управления качеством и поддержку постоянного улучшения;
xiii) обеспечение своевременного и эффективного обмена информацией и доведение проблемных вопросов по качеству до руководящего состава соответствующего уровня.
Обучение
2.10. Производитель должен обеспечить обучение персонала, должностные обязанности которого предполагают пребывание в производственных и складских зонах или контрольных лабораториях (включая технический и обслуживающий персонал и сотрудников, проводящих уборку), а также другого персонала, деятельность которого может оказать влияние на качество продукции.
2.11. Кроме основного обучения, включающего теорию и практику применения системы управления качеством и GMP, каждый принятый на работу сотрудник должен пройти первичное обучение в соответствии со своими должностными обязанностями. Производитель должен проводить последующее непрерывное обучение персонала, периодически оценивая его эффективность на практике. Обучение персонала должно проводиться по программам, утвержденным соответственно руководителем производства либо руководителем подразделения контроля качества. Производитель обязан хранить записи о проведении обучения.
2.12. Персонал, работающий в зонах, где контаминация представляет опасность (например, в чистых зонах или в зонах, где работают с высокоактивными, токсичными, инфицирующими или сенсибилизирующими веществами), должен пройти специальное обучение.
2.13. Посетители и (или) не прошедшие обучение сотрудники не должны допускаться в зоны производства и контроля качества. Если это неизбежно, они должны предварительно пройти инструктаж, в частности по гигиеническим требованиям к персоналу и использованию защитной одежды. Должны быть организованы их сопровождение и наблюдение за ними.
2.14. При обучении должны подробно разъясняться и обсуждаться принципы фармацевтической системы качества, а также все меры, улучшающие их понимание и осуществление.
Гигиенические требования к персоналу
2.15. На предприятии должны быть разработаны детальные программы по гигиене труда с учетом особенностей конкретного производства. Эти программы должны содержать процедуры, касающиеся здоровья, соблюдения гигиенических правил и требований к одежде персонала. Каждый сотрудник, обязанности которого предполагают пребывание в зонах производства и контроля, должен понимать и точно соблюдать эти процедуры. Руководство предприятия должно содействовать развитию программ по гигиене, которые следует обсуждать при обучении.
2.16. Лица, принимаемые на работу, должны пройти медицинский осмотр. Производитель обязан утвердить инструкции, обеспечивающие его осведомленность о состоянии здоровья персонала, которое может повлиять на качество продукции. После первичного медицинского осмотра должны проводиться регулярные последующие медицинские осмотры персонала.
2.17. Производитель должен принять меры, обеспечивающие недопущение лиц с инфекционными заболеваниями или открытыми повреждениями на открытых участках тела к производству лекарственных средств.
2.18. Лица, входящие в производственные зоны, должны носить защитную одежду, соответствующую выполняемым в этих зонах операциям.
2.19. В производственных и складских зонах запрещаются курение, прием пищи, питье, жевание, а также хранение пищевых продуктов, напитков, табачных изделий и личных лекарственных препаратов. Не допускаются любые действия, нарушающие гигиенические требования в производственных помещениях (зонах) или других местах, которые могут оказать неблагоприятное влияние на качество продукции.
2.20. Необходимо избегать непосредственного контакта рук персонала с открытой продукцией, а также с любой частью оборудования, контактирующей с продукцией.
2.21. Персонал должен быть обучен правилам мытья рук.
2.22. Специальные требования, относящиеся к производству отдельных видов продукции (например, стерильных лекарственных средств), приведены в приложениях к настоящим Правилам.
Консультанты
2.23. Консультанты должны иметь соответствующее образование, подготовку и опыт по вопросам, касающимся деятельности, для консультирования в сфере которой они привлекаются. Следует вести их учет с указанием личных данных, адреса проживания, квалификации и вида услуг, предоставляемых этими консультантами.
Глава 3. Помещения и оборудование Принцип
Помещения и оборудование следует располагать, проектировать, строить, оснащать и эксплуатировать таким образом, чтобы они соответствовали проводимым операциям. Их расположение и конструкция должны сводить к минимуму риск ошибок и обеспечивать возможность эффективной очистки и обслуживания в целях исключения перекрестной контаминации, накопления пыли или грязи и любых неблагоприятных факторов для качества продукции.
Помещения Общие требования
3.1. Производственная среда помещений, учитывая все меры по защите производства, должна представлять минимальный риск контаминации материалов или продукции.
3.2. Следует проводить тщательное техническое обслуживание помещений, гарантируя, что ремонт и обслуживание не будут представлять никакой опасности для качества продукции. Помещения следует убирать и, где применимо, дезинфицировать в соответствии с подробными письменными инструкциями.
3.3. Освещение, температура, влажность и вентиляция должны быть соответствующими и не оказывать неблагоприятного воздействия (прямого или косвенного) ни на лекарственные препараты во время их производства и хранения, ни на надлежащее функционирование оборудования.
3.4. Помещения должны быть спроектированы и оснащены таким образом, чтобы обеспечивать максимальную защиту от проникновения в них насекомых и (или) животных.
3.5. Должны быть приняты меры, предотвращающие вход в помещения лиц, не имеющих права доступа в них. Зоны производства, хранения и контроля качества не должны использоваться как проходные для персонала, который в них не работает.
Производственная зона
3.6. Перекрестная контаминация должна быть предотвращена для всех лекарственных средств при проектировании и эксплуатации производственных помещений. Меры по предотвращению перекрестной контаминации должны быть соизмеримы с рисками. Для оценки и управления рисками следует использовать принципы управления рисками для качества. В зависимости от уровня риска могут потребоваться выделенные помещения и оборудование для производственных и (или) упаковочных операций, чтобы контролировать риск возможной перекрестной контаминации некоторыми лекарственными средствами.
Необходимо выделять производственные участки, если лекарственное средство представляет собой риск:
i) который не может контролироваться надлежащим образом организационными и (или) техническими мерами, или
ii) научные данные токсикологической оценки не подтверждают возможность надлежащего контроля риска (например, высокосенсибилизирующие материалы с аллергенным потенциалом, такие как бета-лактамы), или
iii) соответствующие пределы остаточных количеств, полученные путем токсикологической оценки, не могут быть удовлетворительно определены с помощью валидированного аналитического метода.
Дальнейшие указания приведены в главе 5 настоящих Правил и в приложениях № 2 - 6 к настоящим Правилам.
3.7. Предпочтительно, чтобы планировочные решения помещений соответствовали логической последовательности производственных операций и требуемым уровням чистоты.
3.8. Планировочные решения рабочих зон и внутрипроизводственных зон хранения должны обеспечивать последовательное и логичное размещение оборудования и материалов, сводящее к минимуму риск перепутывания различных лекарственных препаратов или их компонентов, обеспечивающее отсутствие перекрестной контаминации и сводящее к минимуму риск пропуска или неправильного осуществления любого этапа при производстве или контроле.
3.9. Там, где исходное сырье и первичные упаковочные материалы, промежуточная или нерасфасованная продукция подвержены влиянию производственной среды, внутренние поверхности (стены, полы и потолки) должны быть гладкими, без щелей и трещин на стыках, не должны выделять частиц, а также должны легко и эффективно очищаться и, при необходимости, дезинфицироваться.
3.10. Трубопроводы, осветительные приборы, вентиляционные установки и другие системы обслуживания должны быть спроектированы и расположены таким образом, чтобы не было углублений, затрудняющих их очистку. По возможности доступ к ним для обслуживания должен осуществляться извне производственных зон.
3.11. Точки подключения к канализационным стокам должны быть соответствующих размеров и оборудованы устройствами для предотвращения обратного потока. По возможности следует избегать открытых сливных желобов, но если они необходимы, то они должны быть неглубокими для облегчения очистки и дезинфекции.
3.12. Производственные зоны следует эффективно вентилировать; в них должны быть средства для контроля параметров воздуха (включая температуру и, где необходимо, влажность и фильтрацию), соответствующие обрабатываемой продукции, проводимым операциям и производственной зоне.
3.13. Взвешивание исходного сырья, как правило, следует осуществлять в отдельном, предназначенном для этого помещении.
3.14. В тех случаях, когда происходит образование пыли (например, во время отбора проб, взвешивания, смешивания и производственных операций, упаковки сухой продукции), должны быть приняты специальные меры предосторожности в целях предупреждения перекрестной контаминации и облегчения очистки.
3.15. Помещения для упаковки лекарственных препаратов должны быть специально спроектированы и расположены таким образом, чтобы избежать перепутывания или перекрестной контаминации.
3.16. Производственные зоны должны быть хорошо освещены, особенно там, где проводится постоянный визуальный контроль.
3.17. Контроль в процессе производства можно проводить в производственной зоне, если это не создает риска для технологического процесса.
Складские зоны
3.18. Складские зоны должны быть достаточно вместительными, чтобы обеспечить упорядоченное хранение различных категорий материалов и продукции: исходного сырья и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции, а также продукции, находящейся в карантине, продукции, разрешенной для выпуска, отклоненной, возвращенной или отозванной.
3.19. При проектировании и оснащении складских зон следует предусматривать надлежащие условия хранения. В частности, они должны быть чистыми и сухими, в них должна поддерживаться требуемая температура. Если требуются специальные условия хранения (например, температура, влажность), то необходимо обеспечивать и проверять такие условия, а также осуществлять их мониторинг.
3.20. В местах приемки и отгрузки должна быть обеспечена защита сырья, материалов и продукции от воздействия погодных условий. Зоны приемки должны быть спроектированы и оборудованы так, чтобы тару с поступающим сырьем и материалами перед складированием при необходимости можно было очищать.
3.21. Если режим карантина обеспечивается хранением продукции в раздельных зонах, то эти зоны должны быть четко обозначены, а доступ в них разрешен персоналу, имеющему соответствующие полномочия. Любая другая система, заменяющая физический карантин, должна обеспечивать эквивалентную надежность.
3.22. Как правило, должна быть отдельная зона для отбора проб исходного сырья. Если отбор проб осуществляется в зоне хранения, то он должен проводиться таким образом, чтобы предотвратить контаминацию или перекрестную контаминацию.
3.23. Для хранения отклоненных, отозванных или возвращенных сырья, материалов или продукции должны быть предусмотрены изолированные зоны.
3.24. Высокоактивные вещества и лекарственные препараты должны храниться в безопасных и защищенных зонах.
3.25. Следует уделять особое внимание безопасному и надежному хранению печатных упаковочных материалов, так как они считаются критическими для обеспечения соответствия лекарственного препарата установленным требованиям.
Зоны контроля качества
3.26. Как правило, лаборатории контроля качества должны быть отделены от производственных зон. Это особенно важно для лабораторий по контролю биологических и микробиологических лекарственных препаратов и радиоизотопов, которые должны быть также отделены друг от друга.
3.27. Контрольные лаборатории должны быть спроектированы таким образом, чтобы соответствовать требованиям к проводимым в них работам. Во избежание перепутывания и перекрестной контаминации они должны иметь достаточную площадь. Необходимо выделить соответствующие и подходящие площади для хранения образцов и записей.
3.28. Для чувствительных приборов, нуждающихся в защите от вибрации, электромагнитных полей, влажности воздуха и т. д., могут быть предусмотрены отдельные комнаты.
3.29. Особые требования предъявляются к лабораториям, в которых проводятся работы со специфическими веществам (например, биологическими или радиоактивными материалами).
Вспомогательные зоны
3.30. Комнаты отдыха и приема пищи должны быть отделены от других зон.
3.31. Помещения для переодевания, туалеты и душевые кабины должны иметь удобный доступ, их планировка и размеры должны соответствовать численности персонала. Не допускается, чтобы туалеты непосредственно сообщались с производственными или складскими зонами.
3.32. Мастерские по возможности должны быть отделены от производственных зон. В том случае, если хранение запасных частей и инструментов осуществляется в производственной зоне, их следует содержать в предусмотренных для этого комнатах или запирающихся ящиках.
3.33. Виварии должны быть изолированы от других зон, иметь отдельный вход (доступ к животным) и отдельные системы воздухоподготовки.
Оборудование
3.34. Конструкция, монтаж и порядок технического обслуживания производственного оборудования должны соответствовать его назначению.
3.35. Работы по ремонту и техническому обслуживанию оборудования не должны представлять никакой опасности для качества продукции.
3.36. Конструкция производственного оборудования должна быть такой, чтобы его можно было легко и тщательно очищать. Очистку следует проводить в соответствии с подробными письменными инструкциями. Оборудование следует хранить только в чистом и сухом состоянии.
3.37. Инвентарь для мытья и очистки следует выбирать и использовать так, чтобы он не стал источником контаминации.
3.38. Оборудование должно быть установлено таким образом, чтобы не допускать возникновения какого-либо риска ошибок или контаминации.
3.39. Производственное оборудование не должно представлять никакой опасности для продукции. Части производственного оборудования, контактирующие с продукцией, не должны вступать с ней в реакцию, выделять или абсорбировать вещества в такой степени, чтобы это могло повлиять на качество продукции и создать таким образом какую-либо опасность.
3.40. Точность и рабочий диапазон весов и других средств измерений должны соответствовать производственным и контрольным операциям, в которых они используются.
3.41. Калибровка и поверка весов и других средств измерений, регистрирующих и контрольных приборов должна проводиться с определенной периодичностью соответствующими методами. Необходимо оформлять и сохранять записи таких испытаний.
3.42. Стационарные трубопроводы должны иметь четкую маркировку с указанием проходящих по ним веществ и, если требуется, направлений потока.
3.43. Трубопроводы для воды очищенной, воды для инъекций (дистиллированной, деионизированной) и, при необходимости, другой воды следует подвергать санитарной обработке в соответствии с письменными инструкциями, в которых указаны пределы микробной контаминации и принимаемые меры в случае их превышения.
3.44. Неисправное оборудование должно быть удалено из производственных зон и зон контроля качества или промаркировано как неисправное.
Глава 4.
Документация
Принцип
Надлежащая документация составляет неотъемлемую часть системы обеспечения качества и является ключевым элементом работы в соответствии с настоящими Правилами. В системе управления качеством производителя должны быть четко установлены различные виды используемой документации и носителей информации. Документация может существовать в различных формах, в том числе на бумажном, электронном или фотографическом носителе. Главной целью применяемой системы документации должно быть создание, управление, контроль и регистрация всей деятельности, которая может непосредственно или опосредовано влиять на все аспекты качества лекарственных препаратов. В дополнение к надлежащему документальному оформлению различных процессов и оценки каких-либо наблюдений система управления качеством должна содержать достаточно подробные указания. Эти указания способствуют общему пониманию требований таким образом, чтобы можно было продемонстрировать их постоянное соблюдение.
Существует 2 основных вида документации для выполнения требований настоящих Правил и регистрации их соблюдения: регламентирующий - инструкции (указания, требования) и регистрирующий - записи (отчеты). Необходимо применять соответствующую надлежащую практику документального оформления в зависимости от вида документа.
Должен быть внедрен соответствующий контроль для обеспечения точности, целостности, доступности и четкости документов. Регламентирующие документы должны быть доступны в письменном виде и не должны содержать ошибок. Понятие «в письменном виде» используется в значении «записанный или задокументированный на носителях информации, с которых данные могут быть получены в читаемой форме».
Документация, требуемая надлежащей производственной практикой Досье производственной площадки: документ, в котором описана деятельность производителя, имеющая отношение к настоящим Правилам.
Типы регламентирующих документов (руководства и требования):
спецификации - документы, содержащие подробные требования, которым должны соответствовать исходные и упаковочные материалы и продукция, использующиеся или получаемые при производстве. Они являются основой для оценки качества лекарственных препаратов;
производственные рецептуры, технологические инструкции, инструкции по упаковке, методики испытаний - документы, содержащие подробную информацию обо всем используемом исходном сырье, оборудовании и компьютеризированных системах (при их наличии). В этих документах должны содержаться все инструкции по осуществлению технологических процессов, упаковке, отбору проб и проведению испытаний. Где применимо, следует указать все точки контроля в процессе производства, а также используемые процессно-аналитические технологии вместе с критериями приемлемости;
процедуры (стандартные операционные процедуры (далее - СОП)) - документы, содержащие требования к выполнению определенных операций;
протоколы - документы, содержащие требования к проведению и регистрации отдельных операций;
технические соглашения - соглашения, заключенные между заказчиками и исполнителями относительно работ, которые выполняются сторонними организациями (аутсорсинг).
Типы регистрирующих документов (записи (отчеты)): записи - свидетельства, подтверждающие выполнение различных действий для доказательства соответствия инструкциям (например, мероприятий, происшествий, расследований). для произведенных серий также содержат историю каждой серии продукции, включая информацию о ее реализацию. Записи содержат исходные данные, используемые для формирования других записей. В случае использования электронных записей определять, какие данные следует использовать в качестве исходных, должны установленные пользователи. Все данные, на которых основываются решения по качеству, должны определяться в качестве исходных данных. Записи, относящиеся к конкретной серии, могут быть собраны в досье на серию;
сертификаты анализа - документы (паспорта, аналитические листки, другие документы), содержащие резюме результатов испытаний образцов продукции или материалов вместе с оценкой соответствия установленной спецификации. Оценка соответствия серии утвержденному регистрационному досье также может быть основана (целиком или частично) на анализе данных, параметров и результатов, полученных в реальном времени (резюме и отчеты об отклонениях). Такой подход применим, если при производстве серии используется процессно-аналитическая технология (PAT);
отчеты - документы, отражающие выполнение конкретных заданий, проектов или расследований вместе с результатами, выводами и рекомендациями.
Управление документацией
4.1. Должны быть установлены требования ко всем видам документов, и их следует соблюдать. Требования применяются в равной мере ко всем формам документов на различных видах носителей информации. Для сложных систем требуется хорошо задокументированное пояснение, валидация и адекватный контроль. Документы могут быть смешанными по форме, например, некоторые элементы в электронном виде, а другие - на бумажном носителе. Необходимо установить взаимосвязи и средства управления в отношении оригиналов документов, официальных копий, обработки данных и записей, как для смешанных, так и для однотипных систем документации. Необходимо внедрить соответствующие средства управления в отношении таких электронных документов, как шаблоны, формы и первичные документы. Должны быть соответствующие меры контроля для обеспечения целостности записей в течение срока хранения.
4.2. Документы должны тщательно разрабатываться, подготавливаться, согласовываться и распределяться. В зависимости от вида они должны отвечать требованиям соответствующих частей спецификаций исследуемого препарата, регистрационного досье, а также документов, подаваемых для получения лицензии на производство. Копирование оригинальных документов с целью получения рабочих документов не должно допускать возникновение каких-либо ошибок при копировании.
4.3. Регламентирующие документы должны быть утверждены и подписаны лицами, имеющими право подписи, с указанием даты. Содержание документов должно быть однозначным, документы должны иметь уникальную идентификацию. Должна быть определена дата введения в действие.
4.4. Регламентирующие документы должны иметь логичную структуру, обеспечивающую простоту их проверки. Стиль изложения документов должен соответствовать их предполагаемому использованию. Стандартные операционные процедуры, рабочие инструкции и методики должны быть написаны в форме, предполагающей обязательность их выполнения.
4.5. Документы в рамках системы управления качеством следует регулярно пересматривать и актуализировать, необходимо исключить использование устаревших версий.
4.6. Документы не должны оформляться в рукописном виде, однако если предусмотрено рукописное внесение в документы данных, то должно быть достаточно места для таких записей.
Правила надлежащего документального оформления
4.7. Рукописные записи должны быть сделаны четко, разборчиво и так, чтобы внесенные данные нельзя было удалить.
4.8. Записи следует вести при выполнении каждого действия и таким образом, чтобы можно было проследить всю значимую деятельность, касающуюся производства лекарственных препаратов.
4.9. Любое изменение, вносимое в документ, должно быть подписано и датировано. Изменение должно давать возможность прочтения первоначальной информации. Где применимо, должна быть указана причина изменения.
Хранение документов
4.10. Должно быть четко определено, какая запись относится к каждому виду производственной деятельности, и где она находится. Должны быть обеспечены надежные меры контроля, валидированные в соответствующих случаях, для обеспечения целостности записи на протяжении срока хранения.
4.11. Особые требования предъявляются к документации серии, которую следует хранить в течение 1 года после окончания срока годности этой серии или не менее 5 лет после выдачи разрешения на реализацию серии уполномоченным лицом в зависимости от того, какой срок дольше. Для лекарственных препаратов, предназначенных для клинических исследований, документацию серии следует хранить как минимум 5 лет после окончания или официального прекращения последних клинических исследований, в которых использовали эту серию. В требованиях законодательства государств-членов и права Союза к специфическим видам лекарственных препаратов (например, высокотехнологичные лекарственные препараты для наиболее современных видов лечения) могут быть установлены более длительные периоды хранения определенных документов.
4.12. Для других видов документации срок хранения зависит от видов деятельности, которые эта документация сопровождает. Критическую документацию, включая исходные данные (например, касающиеся валидации или стабильности), подтверждающие информацию регистрационного досье, необходимо хранить на протяжении срока действия регистрационного удостоверения.
Допустимо удалять определенную документацию (например, исходные данные, сопровождающие отчеты по валидации или стабильности), если данные были заменены полным комплектом новых данных. Обоснование таких действий должно быть оформлено документально. При этом необходимо учитывать требования к хранению документации серии (например, в случае данных по валидации процесса сопровождающие исходные данные следует хранить по крайней мере такое же время, как и документацию на все серии, для которых разрешение на выпуск подтверждено данными этих валидационных исследований).
В следующих разделах приведены примеры необходимых документов. В системе управления качеством должны быть описаны все документы, необходимые для гарантии качества продукции и безопасности пациентов.
Спецификации
4.13. Необходимо иметь в наличии соответствующим образом утвержденные спецификации на исходные и упаковочные материалы и готовую продукцию с указанием даты утверждения.
Спецификации на исходные и упаковочные материалы
4.14. Спецификации на исходные материалы, первичные или печатные упаковочные материалы должны содержать следующую информацию (соответствующие ссылки на информацию, где применимо):
a) описание материалов, включающее в себя:
наименование и внутренний код;
ссылку на фармакопейную статью или на другую нормативную документацию или нормативный документ (при их наличии);
наименование утвержденных поставщиков и, если это возможно, производителя исходных и упаковочных материалов;
образец печатных материалов;
b) инструкции по отбору проб и проведению испытаний;
c) качественные и количественные показатели с указанием допустимых пределов;
d) условия хранения и меры предосторожности;
e) срок годности или максимальный срок хранения до повторного контроля.
Спецификации на промежуточную и нерасфасованную продукцию
4.15. Спецификации на промежуточную и нерасфасованную продукцию должны быть в наличии для критических стадий или при ее приобретении и отгрузке. Эти спецификации должны быть аналогичны спецификациям соответственно на исходное сырье либо готовую продукцию.
Спецификации на готовую продукцию
4.16. Спецификации на готовую продукцию должны содержать следующие данные:
a) наименование лекарственного препарата и код (при необходимости);
b) состав или ссылка на соответствующий документ;
c) описание лекарственной формы и подробные сведения об упаковке;
d) инструкции по отбору проб и проведению испытаний или ссылка на соответствующий документ;
e) качественные и количественные показатели с указанием допустимых пределов;
f) условия хранения и особые меры предосторожности при использовании (при необходимости);
g) срок годности.
Производственная рецептура и технологические инструкции
На каждый производимый лекарственный препарат и каждый размер серии необходимо иметь утвержденные письменные производственную рецептуру и технологические инструкции.
4.17. Производственная рецептура должна включать в себя:
a) наименование лекарственного препарата со ссылкой на код в соответствии со спецификацией;
b) описание лекарственной формы, дозировки препарата и размера серии;
c) перечень всех исходных материалов, которые будут использоваться, с указанием количества каждого. Также должны быть указаны все вещества, которые могут исчезнуть в ходе технологического процесса;
d) ожидаемый выход готовой продукции с указанием допустимых пределов и выходы соответствующих промежуточных продуктов, где это возможно.
4.18. Технологические инструкции должны содержать:
a) данные о месте осуществления процесса и об основном оборудовании, которое должно при этом использоваться;
b) методы или ссылки на методы, которые должны использоваться для подготовки критического оборудования (например, очистка, монтаж, калибровка, стерилизация);
c) инструкции по проверке того, что оборудование и рабочее место свободны от предыдущей продукции, ненужных для запланированного процесса документов и материалов, а также по проверке чистоты оборудования и его готовности к следующему процессу;
d) подробные постадийные технологические инструкции (например, проверка материалов, предварительная обработка, порядок загрузки сырья, критические параметры процесса (время, температура и т. п.));
e) инструкции по всем видам контроля в процессе производства с указанием допустимых пределов;
f) требования к хранению нерасфасованной продукции при необходимости, включая требования к таре, маркировке и специальным условиям хранения, где это требуется;
g) все подлежащие соблюдению особые меры предосторожности.
Инструкции по упаковке
4.19. Для каждого лекарственного препарата, размера и типа упаковки должны быть в наличии инструкции по упаковке. Как правило, они должны включать в себя следующие сведения (ссылки на них):
a) наименование лекарственного препарата, включая номер серии нерасфасованной продукции и готового продукта;
b) описание его лекарственной формы и дозировки (где применимо);
c) количество лекарственного препарата в окончательной упаковке, выраженное в штуках, единицах массы или объема;
d) полный перечень всех необходимых упаковочных материалов, включая их количества, размеры и типы с указанием кода или номера, относящихся к спецификациям на каждый упаковочный материал;
e) где применимо, образец или копия соответствующих печатных упаковочных материалов и образцы, указывающие на место нанесения номера серии и срока годности продукции;
f) требования по проверке того, что оборудование и рабочее место свободны от предыдущей продукции, документов или материалов, ненужных для запланированных операций по упаковке (очистка линии), а также по проверке чистоты оборудования и его готовности к следующему процессу;
g) сведения о подлежащих соблюдению специальных мерах предосторожности (включая тщательную проверку зоны упаковки и оборудования), гарантирующих очистку упаковочной линии перед началом работы;
h) описание процесса упаковки со всеми основными вспомогательными операциями и используемым оборудованием;
i) описание контроля в процессе производства с указаниями по отбору проб и допустимых пределов.
Дополнительно могут быть разработаны иные документы, конкретизирующие положения производственной рецептуры и технологических инструкций.
Записи по производству серии
4.20. На каждую произведенную серию следует сохранять записи по производству серии.
Они должны основываться на соответствующих частях утвержденных документов (производственной рецептуры и технологических инструкций) и содержать следующую информацию:
a) наименование и номер серии продукции;
b) даты и время начала и завершения технологического процесса, а также основных промежуточных стадий;
c) фамилия и инициалы оператора (операторов) каждой основной технологической операции, а также лица, проверившего каждую из этих операций, при необходимости;
d) номер серии и (или) номер аналитического контроля, а также фактически отвешенное количество исходных материалов каждого вида (включая номер серии и количество любого добавленного регенерированного или переработанного материала);
e) сведения о любой относящейся к делу технологической операции или любом действии, а также об основном использованном оборудовании;
f) записи по контролю в процессе производства с указанием исполнителей и полученных результатов;
g) выход продукции на различных стадиях производства;
h) сведения об особых проблемах с подписанным разрешением на любое отклонение от технологических инструкций;
i) подпись лица, ответственного за технологический процесс, с указанием даты.
В случае если валидированный процесс подвергается постоянному мониторингу и контролю, автоматически создаваемые отчеты могут ограничиваться кратким резюме о соответствии и отчетами об отклонениях (отступлениях) от спецификации.
Записи по упаковке серии
4.21. На каждую произведенную серию или часть серии следует сохранять записи по упаковке серии. Они должны основываться на соответствующих частях инструкций по упаковке.
Записи по упаковке серии должны включать в себя следующие данные:
a) наименование и номер серии лекарственного препарата;
b) дата (даты) и время проведения операций по упаковке;
c) фамилия и инициалы оператора (операторов) каждой основной технологической операции, а также лица, проверившего каждую из этих операций, при необходимости;
d) записи проверок идентичности и соответствия инструкциям по упаковке, включая результаты контроля в процессе производства;
e) подробные сведения об осуществленных операциях по упаковке, включая ссылки на использованное оборудование и упаковочные линии;
f) образцы использованного печатного упаковочного материала, включая образцы с нанесенными номером серии, сроком годности и прочими дополнительными маркировочными данными, где применимо;
g) сведения об особых проблемах или необычных происшествиях с подписанным разрешением на любое отклонение от инструкций по упаковке;
h) количество и ссылка на номер или наименование всех печатных упаковочных материалов и нерасфасованной продукции, выданных, использованных, уничтоженных или возвращенных на склад, а также количество готового продукта для составления соответствующего баланса. Электронный контроль в процессе упаковки является основанием для невключения такой информации;
i) подпись лица, ответственного за процесс упаковки, с указанием даты.
Процедуры и записи
Приемка
4.22. На приемку каждой поставки каждого вида исходных материалов (в том числе нерасфасованной, промежуточной или готовой продукции), а также первичных, вторичных и печатных упаковочных материалов должны быть в наличии письменные процедуры и подтверждающие записи.
4.23. Записи по приемке должны содержать:
a) наименование материала в накладной и на таре;
b) внутризаводское наименование (если оно отличается от наименования, указанного в подпункте «а» настоящего пункта) и (или) код материала (при необходимости);
c) дату приемки;
d) наименование поставщика и наименование производителя;
e) номер серии производителя или кодовый номер;
f) общее количество полученных материалов и число единиц упаковок;
h) номер серии, присвоенный после приемки;
i) любые существенные замечания.
4.24 Должны быть в наличии письменные процедуры по внутризаводской маркировке, карантину и хранению исходных, упаковочных и, если необходимо, других материалов.
Отбор проб
4.25. Должны быть в наличии письменные процедуры по отбору проб, содержащие сведения об используемых методах и оборудовании, количествах, которые должны быть отобраны, и любых подлежащих соблюдению мерах предосторожности во избежание контаминации материала или любого ухудшения его качества.
Проведение испытаний
4.26. Должны быть в наличии письменные методики испытания образцов материалов и продукции на различных стадиях производства с указанием используемых методов и оборудования. Проведенные испытания должны быть документально оформлены.
Прочее
4.27. Должны быть в наличии письменные процедуры, устанавливающие порядок выпуска и отклонения материалов и продукции, в частности выдачи уполномоченным лицом (лицами) разрешения на выпуск готовой продукции. Все записи должны быть доступны уполномоченному лицу. Должна быть внедрена система для обозначения специальных наблюдений и любых изменений в отношении критических данных.
4.28. Следует вести и сохранять записи по реализации каждой серии продукции в целях облегчения отзыва этой серии в случае необходимости.
4.29. Должны быть в наличии письменно изложенные политики, принципы, процедуры, планы, протоколы, отчеты и относящиеся к ним записи предпринятых действий или сделанных заключений, где применимо, в отношении:
валидации и квалификации процессов, оборудования и систем;
монтажа и калибровки оборудования;
переноса технологий;
технического обслуживания, очистки и дезинфекции;
персонала, включая списки лиц с образцами подписей, обучение настоящим Правилам и техническим вопросам, переодеванию и гигиеническим требованиям, а также проверку эффективности обучения;
мониторинга производственной среды;
мероприятий, направленных на осуществление контроля появления и распространения вредителей;
претензий;
отзывов продукции;
возвратов продукции; контроля изменений;
расследования отклонений и несоответствий;
внутреннего аудита качества (соответствия требованиям настоящих Правил);
обобщения записей (например, обзор качества продукции) при необходимости;
аудита поставщиков.
4.30. Должны быть в наличии четкие инструкции по эксплуатации основных единиц производственного и контрольно-аналитического оборудования.
4.31. Необходимо вести регистрационные журналы для наиболее важного или критического технологического и контрольно-аналитического оборудования, а также для помещений, где производилась продукция. В этих журналах следует регистрировать в хронологическом порядке любое использование этих помещений, оборудования и методов, проведение калибровки, технического обслуживания, очистки или ремонта с указанием дат и лиц, выполнивших эти работы.
4.32. Следует вести учет документов в рамках системы управления качеством.
Глава 5. Производство Принцип
Технологические операции должны осуществляться по четко установленным процедурам, они должны отвечать настоящим Правилам для получения продукции требуемого качества и соответствовать разрешению (лицензии) на производство и регистрационному досье.
Общие требования
5.1. Производственный процесс должен осуществляться и контролироваться квалифицированным персоналом.
5.2. Все действия, проводимые с материалами и продукцией, такие как приемка и карантин, отбор проб, хранение, маркировка, выдача в производство, технологический процесс, упаковка и реализация, следует осуществлять согласно письменным процедурам или инструкциям и оформлять документально.
5.3. Все поступающие материалы должны быть проверены, чтобы гарантировать, что поставка соответствует заказу. Тара должна быть очищена (при необходимости) и маркирована с указанием требуемой информации.
5.4. Факты повреждения тары и упаковки и любые другие проблемы, которые могут неблагоприятно повлиять на качество материалов, должны быть расследованы, оформлены документально, а информация о них должна быть доложена в подразделения контроля качества.
5.5. Поступающие материалы и произведенная готовая продукция должны немедленно помещаться в карантин, организованный по принципу раздельного хранения или за счет организационных мер, и содержаться в нем до получения разрешения на их использование или реализацию.
5.6. Приемку закупаемых промежуточной и нерасфасованной продукции проводят в соответствии с правилами, действующими для исходных материалов.
5.7. Все материалы и продукцию следует хранить в соответствующих условиях, установленных их производителем, в определенном порядке, обеспечивающем разделение по сериям и установленную очередность использования складских запасов.
5.8. Следует проводить проверки выходов и материального баланса, чтобы убедиться в отсутствии расхождений с допустимыми предельными значениями.
5.9. Не допускается одновременное или последовательное проведение операций с различными продуктами в одном и том же помещении, за исключением случаев, если не существует риска перепутывания или перекрестной контаминации.
5.10. Продукция и материалы должны быть защищены от микробной и другой контаминации на всех стадиях производства.
5.11. При работе с сухими материалами и продукцией необходимо принимать особые меры предосторожности по предотвращению образования и распространения пыли. Это особенно важно при работе с высоко активными и сенсибилизирующими веществами.
5.12. В течение всего процесса производства все используемые материалы, тара для нерасфасованной продукции, основные единицы оборудования и при необходимости помещения должны быть маркированы этикетками или иным способом с указанием производимой продукции или обрабатываемых материалов, а также их дозировки (где применимо) и номера серии. Там, где это приемлемо, такая маркировка должна также указывать стадию технологического процесса.
5.13. Этикетки, прикрепленные к контейнерам, оборудованию или помещениям, должны быть четкими, однозначными, а также соответствовать установленной на предприятии форме. Рекомендуется в дополнение к информации на этикетках для указания статуса (например, «в карантине», «принято», «отклонено», «чистое» и др.) использовать цветовую маркировку.
5.14. Следует контролировать правильность соединения трубопроводов и других частей оборудования, применяемых для транспортировки продукции из одной зоны в другую.
5.15. Не допускаются любые отклонения от инструкций или процедур. Если происходит отклонение, оно должно быть предварительно письменно санкционировано лицом, имеющим соответствующие полномочия, с привлечением (при необходимости) подразделения контроля качества.
5.16. В производственные помещения может входить только персонал, имеющий право доступа в них.
Предотвращение перекрестной контаминации при производстве
5.17. Производство продукции нелекарственного назначения не должно осуществляться в помещениях и на оборудовании, предназначенных для производства лекарственных средств, но, если это обосновано, может быть разрешено, в случае, если приняты меры по предотвращению перекрестной контаминации лекарственных средств в соответствии с мерами, указанными ниже и в главе 3 настоящих Правил.
Производство и (или) хранение таких технических ядов, как пестициды (кроме случаев, когда они используются для производства лекарственных средств) и гербициды, недопустимо в помещениях, используемых для производства и (или) хранения лекарственных средств.
5.18. Должна быть предотвращена контаминация исходных материалов или продукции другими исходными материалами или продукцией. Такой риск случайной перекрестной контаминации, возникающий в результате неконтролируемого распространения пыли, газов, паров, аэрозолей, генетического материала или организмов от активных веществ, других исходных материалов и продуктов в процессе обработки, от остатков на оборудовании и с одежды операторов, должен быть оценен. Степень риска зависит от природы контаминирующего материала и контаминируемой продукции.
Наиболее опасной является контаминация лекарственных средств, предназначенных для инъекций, а также принимаемых в течение длительного времени.
Тем не менее контаминация любой продукции представляет риск для безопасности пациентов в зависимости от характера и степени контаминации.
5.19. Перекрестную контаминацию необходимо предотвращать, прежде всего за счет надлежащего проектирования помещений и оборудования, как указано в главе 3 настоящих Правил. Это должно быть подкреплено соответствующим дизайном процесса и внедрением любых соответствующих технических или организационных мер, в том числе эффективных и воспроизводимых процессов очистки для контроля риска перекрестной контаминации.
5.20. Для оценки и контроля риска перекрестной контаминации производимой продукции должен быть использован процесс управления рисками для качества, включая оценку активности и токсикологическую оценку. Также следует принять во внимание такие факторы, как дизайн (проект) и использование помещений и оборудования, потоки персонала и материалов, микробиологический контроль, физико-химические характеристики активных веществ, параметры процесса, возможности процессов очистки и аналитические возможности в отношении соответствующих пределов, установленных исходя из оценки производимой продукции. Результат процесса управления рисками для качества должен являться основанием для определения необходимости и уровня, до которого помещения и оборудование должны быть выделены для конкретного лекарственного средства или группы лекарственных средств. Уровень выделения может варьироваться от специально выделенных частей, контактирующих с продуктом, до выделения всего производства. Может быть приемлема локализация производственной деятельности в выделенных автономных производственных зонах на многоцелевом участке, где это оправданно.
5.21. Результаты процесса управления рисками для качества должны стать основой для определения уровня технических и организационных мер, необходимых для контроля рисков перекрестной контаминации. Эти меры могут включать, не ограничиваясь этим, следующее.
Технические меры:
i) выделенные производства (помещения и оборудование);
ii) автономные производственные площади, имеющие отдельное технологическое оборудование и отдельные системы вентиляции и кондиционирования воздуха (HVAC). Также может быть желательным изолировать определенные вспомогательные системы от тех, которые используются в других зонах;
iii) дизайн производственного процесса, помещений и оборудования, позволяющий свести к минимуму возможность перекрестной контаминации в процессе обработки, эксплуатации, технического обслуживания и очистки;
iv) использование «закрытых систем» для обработки и передачи материала (продукта) между оборудованием;
v) использование систем с физическим барьером, в том числе изоляторов, как меры по локализации;
vi) контролируемое удаление пыли вблизи источника загрязнения, например через локальные вытяжные устройства;
vii) выделение технологического оборудования, частей, контактирующих с продуктом, или отдельных частей, которые труднее всего очищать (например, фильтры), инструментов для обслуживания;
viii) использование одноразовых технологий;
ix) использование оборудования, спроектированного с учетом облегчения очистки;
x) надлежащее использование воздушных шлюзов и каскада давлений для локализации потенциального содержащегося в воздухе контаминанта в пределах определенной зоны;
xi) сведение к минимуму риска загрязнения, вызванного рециркуляцией или повторным использованием неочищенного или недостаточно очищенного воздуха;
xii) использование систем автоматической очистки на месте с валидированной результативностью;
xiii) разделение зон мойки оборудования, сушки и хранения для общих зон очистки.
Организационные меры:
i) выделение всего производства или автономных производственных площадей на основе кампаний (выделение с разделением во времени) с последующей очисткой с валидированной результативностью;
ii) хранение специальной защитной одежды внутри зон, где обрабатываются продукты с высоким риском перекрестной контаминации;
iii) верификация очистки после выпуска каждого продукта в целях поддержания эффективности подхода управления риском для качества в отношении продукции высокого риска;
iv) верификация очистки поверхностей, не контактирующих с продукцией, и мониторинг воздуха в производственной зоне и (или) прилегающих зонах в зависимости от риска контаминации для подтверждения эффективности мер против контаминации взвешенными частицами или путем механического переноса;
v) специальные меры по обращению с отходами, загрязненными промывными водами и загрязненной одеждой;
vi) регистрация случаев проливания и рассыпания, инцидентов или отклонений от процедур;
vii) разработка процессов очистки для помещений и оборудования таким образом, чтобы процессы очистки сами по себе не представляли риска перекрестной контаминации;
viii) разработка подробных форм для записей в процессе очистки для обеспечения выполнения очистки в соответствии с утвержденными процедурами и использование этикеток статуса очистки оборудования и производственных зон;
ix) использование общих зон очистки при совместимости процессов;
x) надзор за поведением персонала для обеспечения эффективности обучения и соответствия надлежащим мероприятиям процедурного контроля.
5.22. Мероприятия по предотвращению перекрестной контаминации и их эффективность следует периодически проверять в соответствии с установленными процедурами.
Валидация
5.23. Мероприятия по валидации должны способствовать выполнению настоящих Правил и должны проводиться в соответствии с установленными процедурами. Результаты проведенных мероприятий и заключения по ним должны быть оформлены документально.
5.24. При введении новой производственной рецептуры или нового метода производства необходимо доказать их пригодность для серийного производства. Должно быть доказано, что данный процесс при использовании предусмотренных материалов и оборудования позволяет постоянно производить продукцию требуемого качества.
5.25. Существенные изменения производственного процесса, включая любое изменение оборудования или исходных и упаковочных материалов, которое может повлиять на качество продукции и (или) воспроизводимость процесса, должны пройти валидацию.
5.26. Процессы и процедуры следует подвергать периодической ревалидации (повторной валидации) для гарантии того, что они остаются пригодными для достижения определенных результатов.
Исходные материалы
5.27. В рамках фармацевтической системы качества должны быть задокументированы выбор, квалификация, утверждение и поддержание статуса поставщиков исходных материалов наряду с закупками и приемкой. Уровень контроля должен быть пропорционален рискам, связанным с конкретными материалами, с учетом источника их происхождения, производственного процесса, сложности цепи поставки и конечного назначения материала в лекарственном средстве. Для каждого утвержденного поставщика или материала должны быть в наличии подтверждающие свидетельства. Персоналу, вовлеченному в эту деятельность, необходимо иметь актуальные знания о поставщиках, цепях поставок и связанных с ними рисках. По возможности исходные материалы следует приобретать непосредственно у их производителя.
5.28. Требования к качеству исходных материалов, установленные производителем лекарственного препарата, должны быть согласованы с поставщиками. Соответствующие аспекты производства, испытаний и контроля, в том числе требования к обработке, маркировке, упаковыванию и реализации, процедуры по рассмотрению претензий, отзыву и изъятию, должны быть зафиксированы в официальных соглашениях по качеству или в спецификациях.
5.29. Для утверждения и поддержания статуса поставщиков активных фармацевтических субстанций и вспомогательных веществ требуется следующее.
Для активных фармацевтических субстанций: должна быть установлена прослеживаемая цепь поставки. Связанные с цепью поставки риски (от исходных материалов для активной фармацевтической субстанции до готового лекарственного препарата) должны подвергаться формальной оценке и периодической проверке. Должны существовать соответствующие меры по снижению степени риска в отношении качества фармацевтической субстанции;
должны быть в наличии и сохраняться записи о прослеживаемости каждой цепи поставки;
должны быть проведены аудиты производителей и дистрибьюторов фармацевтических субстанций, в целях подтверждения соответствия требованиям надлежащей производственной практики и надлежащей дистрибьюторской практики. Держатель лицензии на производство лекарственного препарата обязан проверять соблюдение таких требований самостоятельно либо через лицо, действующее от его имени по контракту;
для обеспечения оценки соблюдения настоящих Правил аудиты должны иметь соответствующую продолжительность и область аудита. Следует уделить внимание источникам потенциальной перекрестной контаминации от других материалов, используемых на производственной площадке. Отчет должен полностью отражать всю информацию, включая любые обнаруженные в результате аудита недостатки. Должны быть осуществлены все необходимые корректирующие и предупреждающие действия;
последующие аудиты должны проводиться с установленной на основе анализа рисков периодичностью для обеспечения соблюдения стандартов и дальнейшего использования утвержденной цепи поставки.
Для вспомогательных веществ:
вспомогательные вещества и поставщики вспомогательных веществ должны контролироваться на основе результатов формализованной системы оценки рисков для качества.
5.30. В каждой поставке исходных материалов тара должна быть проверена на целостность упаковки, в том числе целостность пломб, а также на соответствие сведений, указанных в накладной, этикеткам поставщика и утвержденной производителем и поставщиком информации, одобренной производителем лекарственного препарата. Приемочные проверки должны быть задокументированы.
5.31. Если одна поставка материала состоит из различных серий, то каждую серию необходимо рассматривать как отдельную в отношении отбора проб, проведения испытания и выдачи разрешения на использование.
5.32. Находящиеся в складской зоне исходные материалы должны быть соответствующим образом маркированы согласно подпункту 5.13 части I настоящих Правил. Этикетки должны содержать следующую информацию:
присвоенное наименование продукции и при необходимости внутризаводской код;
номер серии, присвоенный при получении;
статус содержимого, где применимо (например, в карантине, на испытании, разрешено, отклонено);
срок годности или дата, после которой требуется повторный контроль, где применимо.
Если используются полностью компьютеризированные системы хранения, то указанная информация не обязательно должна содержаться на этикетке в читаемой форме.
5.33. Должны быть определены соответствующие процедуры или меры, гарантирующие подлинность содержимого каждой единицы тары исходных материалов. Тара, из которой были отобраны пробы, должна быть промаркирована согласно подпункту 6.13 части I настоящих Правил.
5.34. Следует использовать только те исходные материалы, использование которых разрешено подразделением контроля качества и срок годности которых еще не истек.
5.35. Производители готовой продукции несут ответственность за все испытания исходных материалов, которые указаны в регистрационном досье. Производители готовой продукции могут использовать частично или полностью результаты испытаний утвержденного производителя исходных материалов, но должны как минимум выполнить испытание на подлинность каждой серии согласно приложению № 8.
5.36. Обоснование передачи испытаний для исполнения сторонней организацией должно быть оформлено документально. Должны быть соблюдены следующие требования:
i) следует уделить особое внимание контролю за распределением (транспортированием, оптовой реализацией, хранением и поставкой) исходных материалов в целях поддержания характеристик качества исходных материалов и обеспечения того, чтобы результаты испытаний были по-прежнему применимы к поставленным материалам;
ii) производитель лекарственного препарата должен проводить аудиты площадки (площадок), осуществляющих испытания исходных материалов (в том числе и отбор проб), как самостоятельно, так и через третьих лиц с периодичностью, определенной с учетом рисков. Это необходимо для гарантии соответствия требованиям надлежащей производственной практики, спецификациям и методам испытаний, описанным в регистрационном досье;
iii) сертификат анализа, представленный производителем (поставщиком) исходных материалов, должен быть подписан назначенным лицом с соответствующей квалификацией и опытом. Подпись гарантирует, что каждая серия была проверена на соответствие согласованным спецификациям исходных материалов, если такое свидетельство не представляется отдельно;
iv) производитель лекарственного препарата должен иметь соответствующий опыт работы с производителем исходных материалов (в том числе через дистрибьютора), включающий оценку ранее полученных от него серий исходных материалов и историю их соответствия, для принятия решения о сокращении объема собственных (внутренних) испытаний. Следует учитывать любые существенные изменения в процессах производства или испытаний;
v) производитель лекарственного препарата должен также осуществлять (самостоятельно или с использованием отдельной утвержденной контрактной лаборатории) полный контроль с периодичностью, определенной с учетом рисков, и сравнивать результаты с сертификатом анализа поставщика или производителя исходных материалов с целью проверки надежности последнего. Если в ходе испытаний выявятся расхождения, то должно быть проведено расследование и приняты соответствующие меры. Сертификаты анализа поставщика или производителя исходных материалов не принимаются до тех пор, пока эти меры не будут завершены.
5.37. Исходные материалы должны выдаваться только специально назначенными лицами в соответствии с документально оформленной процедурой, чтобы гарантировать, что нужные исходные материалы точно отвешены или отмерены в чистую и надлежащим образом маркированную тару.
5.38. Каждый выданный материал, его масса или объем должны подвергаться независимой проверке с записью результатов.
5.39. Исходные материалы, выданные для каждой серии, должны храниться в одном месте и иметь соответствующую маркировку.
Технологические операции: промежуточная
и нерасфасованная продукция
5.40. Перед началом любой технологической операции должны быть приняты меры, гарантирующие, что рабочая зона и оборудование очищены и освобождены от любых исходных материалов, продукции, остатков продукции или документации, не имеющих отношения к запланированной операции.
5.41. Промежуточную и нерасфасованную продукцию следует хранить в надлежащих условиях.
5.42. Критические процессы должны пройти валидацию согласно подпунктам 5.23 - 5.26 части I настоящих Правил.
5.43. Должны быть проведены и оформлены документально необходимый контроль в процессе производства и контроль производственной среды.
5.44. Любое существенное отклонение от ожидаемого выхода продукции должно быть оформлено документально и расследовано.
Упаковочные материалы
5.45. Закупке и контролю первичных и печатных упаковочных материалов, а также обращению с ними следует уделять такое же внимание, как и в случае с исходными материалами.
5.46. Особое внимание следует уделять печатным материалам. Их следует хранить в надежных условиях, исключающих доступ посторонних лиц. Разрезанные этикетки и другие разрозненные печатные материалы должны храниться и транспортироваться раздельно в закрытой таре, исключающей их перепутывание. Разрешение на использование упаковочных материалов должно выдаваться только специально назначенными лицами в соответствии с утвержденной и документально оформленной процедурой.
5.47. Каждой поставке или серии первичных или печатных упаковочных материалов должен быть присвоен идентификационный номер или идентификационный знак.
5.48. Просроченные или непригодные к использованию печатные или первичные упаковочные материалы должны быть уничтожены с документальным оформлением.
Операции по упаковке
5.49. При составлении планов операций по упаковке особое внимание должно быть уделено сведению к минимуму риска перекрестной контаминации, перепутывания или подмены. Не допускается упаковывать продукцию различных видов в непосредственной близости друг от друга за исключением случаев, предусматривающих физическое разделение.
5.50. Перед началом операций по упаковке должны быть предприняты меры, гарантирующие, что рабочая зона, упаковочные линии, печатные машины и другое оборудование являются чистыми и не содержат любые использовавшиеся ранее лекарственные препараты, материалы или документы, если они не требуются для запланированной операции. Очистку линии следует проводить согласно соответствующей процедуре.
5.51. Наименование и номер серии упаковываемой продукции должны быть указаны на каждом упаковочном месте или линии.
5.52. При поступлении продукции и упаковочных материалов на участок упаковки следует проверить их количество, идентичность и соответствие инструкциям по упаковке.
5.53. Материалы первичной упаковки должны быть чистыми до начала операции наполнения. Следует уделять внимание предотвращению и устранению любой контаминации, такой как осколки стекла и металлические частицы.
5.54. Как правило, маркировку следует наносить как можно быстрее после фасовки и укупорки. До нанесения маркировки следует принять необходимые меры, исключающие перепутывание или ошибочную маркировку.
5.55. Правильность выполнения любых печатных операций (нанесения номеров серий, срока годности), осуществляемых либо как отдельная технологическая операция, либо в процессе упаковки, следует тщательно контролировать и оформлять документально. Особое внимание следует уделять ручной маркировке, которую следует регулярно перепроверять.
5.56. Особые меры предосторожности должны соблюдаться при использовании разрезанных этикеток и нанесении штампов вне линии упаковки. Для предотвращения перепутывания печатного материала предпочтительнее использовать этикетки в рулоне вместо разрезанных этикеток.
5.57. Следует проводить проверки, гарантирующие, что все электронные устройства считывания кода, счетчики этикеток и аналогичные устройства работают правильно.
5.58. Маркировка упаковочных материалов, нанесенная с помощью печати или методом тиснения, должна быть отчетливой и устойчивой к выцветанию или стиранию.
5.59. При контроле процесса упаковки продукции на линии следует проверять, как минимум, следующее:
a) общий внешний вид упаковок;
b) комплектность упаковок;
c) использование надлежащих видов продукции и упаковочных материалов;
d) правильность нанесения любой маркировки;
e) правильность работы контрольных устройств на линии.
Образцы, взятые с упаковочной линии, не следует возвращать повторно на линию.
5.60. Если при упаковке продукции возникли непредвиденные обстоятельства, она может быть возвращена в производство только после специальной проверки, проведения расследования и с разрешения лица, имеющего соответствующие полномочия. Указанные действия должны быть оформлены в виде протокола, который следует хранить в установленном порядке.
5.61. При существенном или необычном расхождении, установленном во время составления баланса между количеством нерасфасованной продукции, печатного упаковочного материала и числом произведенных единиц готовой продукции, следует провести расследование и установить причину этого расхождения до выдачи разрешения на выпуск.
5.62. После завершения операций по упаковке любые оставшиеся упаковочные материалы с нанесенным на них номером серии должны быть уничтожены с последующим документальным оформлением. Возврат на склад немаркированных упаковочных материалов производится в соответствии с утвержденной процедурой.
Готовая продукция
5.63. До выдачи разрешения на выпуск готовая продукция должна содержаться в карантине в условиях, установленных производителем.
5.64. До момента получения разрешения на выпуск должна быть проведена оценка готовой продукции и документации в порядке, установленном главой 6 настоящих Правил.
5.65. После выдачи разрешения на выпуск готовая продукция должна храниться как пригодный для реализации запас в условиях, установленных производителем.
Отклоненные, повторно использованные и возвращенные материалы
и продукция
5.66. Отклоненные материалы и продукция должны иметь четкую маркировку и храниться раздельно в зонах с ограниченным доступом. Они подлежат возврату поставщику, переработке (если это допустимо) или уничтожению. Любые выполненные действия должны быть оформлены документально и утверждены лицами, имеющими соответствующие полномочия.
5.67. Переработка отклоненной продукции допускается в исключительных случаях при условии отсутствия ухудшения качества готовой продукции и выполнения всех требований спецификаций. Переработку осуществляют в соответствии с утвержденной процедурой после оценки возможного риска с последующим документальным оформлением.
5.68. Повторное использование всей серии или части ранее произведенных серий соответствующего качества путем объединения с серией такой же продукции на определенной стадии производства должно быть санкционировано заранее. Такое введение следует осуществлять в соответствии с установленной процедурой с учетом оценки возникающих рисков, включая любое возможное влияние на срок годности. Деятельность по повторному использованию следует оформлять документально.
5.69. Необходимость дополнительного контроля любой готовой продукции, прошедшей переработку, или продукции, в которую была включена повторно использованная продукция, определяет подразделение контроля качества.
5.70. Возвращенная с рынка продукция, над которой был утрачен контроль со стороны производителя, должна быть уничтожена, за исключением случаев, когда нет сомнений, что ее качество является удовлетворительным. Решение о повторной реализации, перемаркировке или повторном использовании может быть принято только после критической оценки, проведенной подразделением контроля качества в соответствии с письменной процедурой. При этом необходимо учитывать характер продукции, ее предысторию и состояние, соблюдение специальных условий хранения и время, прошедшее с даты выпуска. При любых сомнениях в отношении качества продукции не допускается ее повторное использование или повторный выпуск, но допускается ее химическая переработка с целью регенерации активных ингредиентов. Все выполняемые действия должны быть оформлены документально.
Нехватка продукции в связи с производственными затруднениями
5.71. Производитель должен сообщать держателю регистрационного удостоверения о любых затруднениях в производственных операциях, которые могут привести к необычному ограничению поставки. Такое сообщение должно осуществляться своевременно для упрощения процедуры уведомления об ограничениях поставки со стороны держателя регистрационного удостоверения, направляемого в адрес уполномоченных органов соответствующих государств-членов.
Глава 6. Контроль качества
Принцип
Положения настоящего раздела должны рассматриваться в сочетании с положениями всех соответствующих разделов настоящих Правил.
Контроль качества распространяется как на процедуры отбора проб, спецификации и на проведение испытаний, так и на процедуры организации, документирования и выпуска, гарантирующие проведение необходимых испытаний, а также обеспечивающие то, что исходные и упаковочные материалы не разрешены для использования, а продукция - для реализации и поставки до тех пор, пока их качество не будет признано соответствующим установленным требованиям.
Контроль качества не ограничивается лабораторными работами и должен быть вовлечен в принятие всех решений, касающихся качества продукции. Основополагающим принципом для удовлетворительной работы подразделения контроля качества считается его независимость от производства.
Общие требования
6.1. У каждого производителя должно быть подразделение контроля качества, независимое от других подразделений и отделов. Руководитель этого подразделения должен иметь соответствующие квалификацию и опыт, в его распоряжении может быть одна или несколько контрольных лабораторий. Подразделение должно быть обеспечено достаточными ресурсами для эффективного выполнения мероприятий по контролю качества.
6.2. Основные обязанности руководителя подразделения контроля качества изложены в главе 2 части I настоящих Правил. Подразделение контроля качества может иметь также и иные обязанности, такие как разработка, валидация и обеспечение выполнения всех процедур по контролю качества, наблюдение за контрольными и (или) архивными образцами материалов и продукции, обеспечение правильной маркировки упаковок с материалами и продукцией, мониторинг стабильности продукции, участие в расследовании претензий в отношении качества продукции и др. Проведение всех операций и их результаты требуют документального оформления.
6.3. Оценка готовой продукции должна охватывать все относящиеся к ней факторы, включая условия производства, результаты испытаний в процессе производства, обзор производственной документации (включая документацию по упаковке), соответствие требованиям спецификаций на готовую продукцию и проверку окончательной упаковки.
6.4. Персонал подразделения контроля качества должен иметь доступ в производственные зоны для осуществления отбора проб и при необходимости проведения расследования.
Надлежащая лабораторная практика контроля качества
6.5. Помещения и оборудование контрольных лабораторий должны отвечать общим и специфическим требованиям, предъявляемым к зонам контроля качества, указанным в главе 3 части I настоящих Правил.
Для предотвращения возможности случайной перекрестной контаминации лабораторное оборудование не должно перемещаться между зонами с высокой степенью риска на рутинной основе. В частности, микробиологическая лаборатория должна быть организована таким образом, чтобы свести к минимуму риск перекрестного загрязнения.
6.6. Персонал, помещения и оборудование контрольных лабораторий должны соответствовать задачам, обусловленным характером и объемом производственных операций. В ряде случаев возможно использование внешних лабораторий при условии выполнения ими требований, указанных в главе 7 части I настоящих Правил, и внесения соответствующих записей в документы по контролю качества.
Документация
6.7. Документация лабораторий должна соответствовать требованиям, указанным в главе 4 части I настоящих Правил. Основная часть этой документации относится к контролю качества. В подразделении контроля качества должна быть доступна следующая документация:
i) спецификации;
ii) процедуры, описывающие отбор проб, проведение испытаний, записи (в том числе аналитические рабочие листы и (или) лабораторные журналы), регистрацию и проверку;
iii) процедуры и записи калибровки и квалификации измерительных приборов и технического обслуживания оборудования;
iv) порядок расследования результатов, имеющих отклонения от спецификаций и выходящих за пределы тенденций (трендов);
v) аналитические отчеты (или) сертификаты анализа или другие документы, подтверждающие качество;
vi) данные мониторинга производственной среды (воздух, вода, другие технологические среды), где они требуются;
vii) записи по валидации методик испытаний, где применимо.
6.8. Любую документацию по контролю качества, связанную с досье на серию лекарственного средства, следует хранить в соответствии с требованиями к сохранению документации серии, предусмотренными главой 4 части I настоящих Правил.
6.9. Для некоторых видов данных (результатов испытаний, выходов, контроля производственной среды и др.) записи должны вестись способом, позволяющим проводить оценку существующих тенденций (трендов). Любые данные с отклонениями от требований спецификации, выходящие за пределы тенденций (трендов), должны быть рассмотрены и направлены для проведения расследований.
6.10. В дополнение к информации, являющейся частью документации серии, должны сохраняться и быть доступными другие исходные данные, зафиксированные в таких документах, как лабораторные журналы и (или) записи.
Отбор проб
6.11. Отбор проб следует осуществлять и документально оформлять в соответствии с документированными процедурами, которые определяют:
метод отбора проб;
используемое оборудование;
количество проб, которое должно быть отобрано;
инструкции по любому требуемому разделению пробы;
тип и состояние контейнера, используемого для проб;
идентификацию контейнеров с отобранными пробами;
любые подлежащие соблюдению особые меры предосторожности, особенно при отборе проб стерильных или вредных веществ;
условия хранения;
инструкции по очистке и хранению оборудования для отбора проб.
6.12. Переданные для испытаний образцы должны быть репрезентативны для серии материала или продукции, из которой они отобраны. Могут быть также отобраны другие пробы для контроля наиболее важных этапов процесса (например, его начала или окончания). Используемый план отбора проб должен быть надлежащим образом обоснован и базироваться на принципах управления рисками.
6.13. Контейнеры с образцами должны иметь этикетку с указанием их содержимого (номер серии), дату отбора проб, а также обозначение тарных мест, из которых были отобраны образцы. Работа с ними должна вестись таким образом, чтобы свести к минимуму риск перепутывания и защитить образцы от неблагоприятных условий хранения.
6.14. Дополнительные требования в отношении контрольных и архивных образцов приведены в приложении № 19 настоящих Правил.
Проведение испытаний
6.15. Методики испытаний должны быть валидированными. Лаборатория, которая использует методику испытаний и которая не выполняла ее первоначальную валидацию, должна верифицировать пригодность методики испытаний. Все операции по проведению испытаний, описанных в соответствующих документах регистрационного досье, следует проводить в соответствии с утвержденными методиками.
6.16. Полученные результаты должны быть документированы. Результаты параметров, определенных в качестве показателей качества или как критические, и все вычисления необходимо проверять на согласованность друг с другом и оценивать для них тенденции (тренды). Все расчеты следует тщательно проверять.
6.17. Проведенные испытания должны быть задокументированы. Записи должны включать в себя следующие данные:
i) наименование исходных материалов или продукции и, где применимо, лекарственной формы;
ii) номер серии и, где применимо, наименование производителя и (или) поставщика;
iii) ссылки на соответствующие спецификации и методики проведения испытаний;
iv) результаты испытания, включая наблюдения и расчеты, и ссылки на все сертификаты анализа;
v) даты проведения испытаний;
vi) фамилии и инициалы лиц, проводивших испытания;
vii) фамилии и инициалы лиц, проверивших проведение испытаний и расчетов (где применимо);
viii) однозначное заключение о выдаче разрешения или отклонении продукции (или решение о другом статусе), дата и подпись ответственного лица;
ix) ссылки на использованное оборудование.
6.18. Весь контроль в процессе производства, включая и тот, который выполняется в производственной зоне производственным персоналом, необходимо осуществлять в соответствии с методиками, утвержденными подразделением контроля качества, а его результаты - документально оформлять.
6.19. Особое внимание следует уделять качеству лабораторных реактивов, мерной посуды, титрованных растворов, стандартных образцов и питательных сред. Их следует готовить и контролировать в соответствии с документированными процедурами. Уровень контроля должен быть соразмерен с их назначением и доступными данными о стабильности.
6.20. Стандартные образцы должны быть признаны пригодными для использования по назначению. Их квалификация и сертификация в качестве таковых должны быть однозначно установлена и документирована. В качестве первичных стандартных образцов предпочтительно использование фармакопейных стандартных образцов из официально признанных источников (при их наличии), если иное не обосновано в полной мере (использование вторичных стандартных образцов разрешено, если была продемонстрирована и документирована их прослеживаемость до первичных стандартных образцов). Эти фармакопейные образцы должны использоваться в целях, описанных в соответствующих фармакопейных статьях (монографиях), если иное не разрешено уполномоченным органом государства-члена.
6.21. Лабораторные реактивы, растворы, стандартные образцы и питательные среды, должны быть маркированы с указанием даты приготовления и, где применимо, вскрытия, и подписи исполнителя. На этикетках должны быть указаны сроки годности реактивов и питательных сред, а также особые условия их хранения. Для титрованных растворов необходимо также указывать дату последнего установления титра и соответствующий последний действующий коэффициент поправки.
6.22. При необходимости на контейнере следует указывать дату получения каждого вещества, используемого для проведения испытаний (например, реактивов, растворов и стандартных образцов). Необходимо соблюдать инструкции по их использованию и хранению. В определенных случаях после получения или перед использованием реактивов может быть необходимо проведение их испытания на идентичность и (или) иного испытания.
6.23. Питательные среды должны быть приготовлены в соответствии с требованиями производителя среды, если иное научно не обосновано. Пригодность всех питательных сред должна проверяться перед их использованием.
6.24. Использованные микробиологические среды и штаммы должны быть подвергнуты деконтаминации в соответствии со стандартной процедурой и утилизированы таким образом, чтобы предотвратить перекрестную контаминацию и сохранение остатков. Сроки хранения используемых микробиологических сред должны быть установлены, документированы и научно обоснованы.
6.25. Животных, используемых для контроля компонентов, материалов или продукции, следует помещать в карантин перед работой с ними. Животных требуется содержать и контролировать таким образом, чтобы обеспечить их пригодность для запланированного использования. Животные должны быть идентифицированы. Необходимо вести соответствующие записи, отражающие историю их использования.
Программа текущего испытания стабильности
6.26. После начала реализации зарегистрированного лекарственного средства следует проводить мониторинг стабильности лекарственного средства согласно действующей на постоянной основе программе, которая позволила бы выявить любую проблему (например, изменения уровней примесей или профиля растворения) со стабильностью продукции данного состава во вторичной (потребительской) упаковке.
6.27. Целями программы текущего изучения стабильности являются осуществление мониторинга продукта на протяжении всего срока его годности и определение того, что продукт остается (и можно ожидать, что останется) в пределах своих спецификаций при хранении в указанных в маркировке условиях.
6.28. Эта программа, главным образом, относится к лекарственному средству в упаковке, предназначенному для реализации, однако следует уделять внимание и нерасфасованной продукции. Например, если нерасфасованную продукцию хранят длительное время перед упаковкой и (или) передачей с производственного участка на участок упаковки, необходимо оценить и изучить влияние таких условий на стабильность упакованной продукции. Дополнительно следует обратить внимание на промежуточную продукцию, которая хранится и используется в течение длительного периода. Исследование стабильности восстановленного продукта (приготовленного перед применением) следует проводить при разработке лекарственного средства, текущий контроль стабильности такой продукции не требуется. Однако, где применимо, может проводиться контроль стабильности восстановленного продукта.
6.29. Программа текущего испытания стабильности должна быть изложена в письменном виде в соответствии с общими правилами, приведенными в главе 4 части I настоящих Правил, а результаты официально представлены в виде отчета. Оборудование, используемое для программы текущего испытания стабильности (в частности, климатические камеры), должно пройти квалификацию и обслуживаться в соответствии с общими правилами, указанными в главе 3 части I настоящих Правил и приложении № 15.
6.30. План и (или) протокол текущего испытания стабильности должен охватывать период до окончания срока годности и содержать следующие данные (но не ограничиваться ими):
i) количество серии (серий) для различных дозировок и разных размеров серий, если это применимо;
ii) соответствующие физические, химические, микробиологические и биологические методы испытаний;
iii) критерии приемлемости;
iv) ссылки на методики испытаний;
v) описание системы контейнера (укупорочный элемент);
vi) частота испытаний (точки контроля во времени);
vii) описание условий хранения;
viii) другие необходимые параметры, специфические для данного лекарственного средства.
6.31. План и (или) протокол текущего испытания стабильности могут иметь отличия от плана первоначального долгосрочного испытания стабильности, представленного в регистрационном досье, при условии обоснования и документирования этого в плане.
6.32. Количество серий и частота испытаний должны обеспечить необходимое количество данных, чтобы иметь возможность провести анализ тенденций. Если не обосновано иное, в программу испытания стабильности ежегодно следует включать как минимум одну серию произведенного лекарственного средства в каждой дозировке и в каждом виде первичной упаковки (исключением являются случаи, когда в течение года не произведено ни одной серии). Если для текущего испытания стабильности лекарственных средств необходимо использование животных и не существует альтернативных валидированных методик, частоту испытаний можно устанавливать с учетом подхода, связанного с оценкой риска. При условии научного обоснования могут быть использованы планы с применением брэкетинга и построения матриц.
6.33. В некоторых случаях в программу текущего испытания стабильности следует включать дополнительные серии. Например, текущее испытание стабильности следует осуществлять после любого значительного изменения или значительного отклонения в процессе производства или упаковывания. Для включения в программу должны быть приняты во внимание любые операции по повторному использованию, переработке или регенерации.
6.34. Результаты текущего испытания стабильности должны быть доступны ключевому персоналу и, в частности, уполномоченному лицу (лицам). Если текущее изучение стабильности проводится на площадке, отличной от производственной площадки, на которой производится нерасфасованный продукт или готовая продукция, то между соответствующими сторонами должно быть заключено письменное соглашение. Результаты текущего испытания стабильности должны представляться на площадку производства для их обзора уполномоченным органом (организацией) государства-члена.
6.35. Результаты, выходящие за пределы спецификации, или существенные нетипичные тенденции (тренды) должны расследоваться. О любом подтвержденном результате, выходящем за пределы спецификации, или существенной негативной тенденции (тренда) необходимо сообщать соответствующим уполномоченным органам или организациям государств-членов. Необходимо рассмотреть возможное влияние на серии, выпущенные на рынок, в соответствии с главой 8 части I настоящих Правил и проконсультироваться с соответствующими уполномоченным органам или организациям государств-членов.
6.36. Следует вести в письменном виде заключения по всем полученным данным, в том числе любые промежуточные выводы по программе. Такие заключения следует подвергать периодическому обзору.
Трансфер (передача) методик испытаний
6.37. До трансфера (передачи) методики испытаний передающая сторона должна убедиться в том, что методика (и) испытания соответствуют описанной в регистрационном досье.
Необходимо провести проверку первоначальной валидации методики испытаний для гарантии соответствия действующим рекомендациям. До начала процесса технического трансфера необходимо провести и документально оформить анализ расхождений, что требуется для определения необходимости проведения каких-либо дополнительных валидационных работ.
6.38. Трансфер (передача) методик испытаний от одной лаборатории (передающей лаборатории) в другую лабораторию (принимающая лаборатория) должен быть описан в подробном протоколе.
6.39. Протокол трансфера (передачи) методик испытаний должен включать следующее:
i) идентификация испытаний, которые должны быть выполнены, и соответствующие методики испытаний, подлежащие передаче;
ii) идентификация дополнительных требований к обучению;
iii) идентификация стандартов и образцов для испытаний;
iv) идентификация любых специальных условий транспортировки и хранения образцов для испытаний;
v) критерии приемлемости, которые должны быть основаны на текущих валидационных исследованиях методологии и связаны с рекомендациями, установленными в рамках Союза.
6.40. Отклонения от протокола должны быть расследованы до завершения процесса трансфера (передачи) методик испытаний. Отчет о трансфере (передаче) методик испытаний должен содержать сравнительный результат процесса и определять области, требующие дальнейшей ревалидации методик испытаний, если применимо.
6.41. Если применимо, необходимо рассмотреть особые требования, описанные в руководствах и касающиеся трансфера конкретных аналитических методик (например, спектроскопии в ближней инфракрасной области).
Глава 7. Деятельность, передаваемая для выполнения
другому лицу(аутсорсинг)
Принцип
Любая деятельность, на которую распространяются настоящие Правила и которая передана другому лицу (аутсорсинг), должна быть надлежащим образом определена, согласована и проконтролирована во избежание разночтений, способных привести к неудовлетворительному качеству продукции или выполняемых работ. Соглашение между заказчиком и исполнителем должно быть оформлено в письменном виде с указанием четко определенных обязанностей каждой из сторон. Система управления качеством заказчика должна точно устанавливать, каким образом уполномоченное лицо, подтверждающее выпуск каждой серии продукции, выполняет свои обязанности в полной мере.
Требования, приведенные в данной главе, устанавливают ответственность производителей перед уполномоченными органами государств-членов в отношении регистрации лекарственных препаратов и лицензирования производства. Они не устанавливают ответственность исполнителя и заказчика перед потребителем, которая регулируется другими нормативно-правовыми актами.
Общие требования
7.1. Деятельность, передаваемая для выполнения другому лицу (аутсорсинг), должна быть оформлена письменным соглашением, в котором указаны продукция, работы или услуги, с которыми связана указанная деятельность, и все связанные с такой деятельностью технические мероприятия.
7.2. Все мероприятия, проводимые в рамках деятельности, переданной для выполнения другому лицу (аутсорсинга), включая любые предложенные изменения технических или иных мероприятий, должны отвечать требованиям законодательства государств-членов и актов, составляющих право Союза, и регистрационному досье лекарственного препарата, где это применимо.
7.3. Если держатель регистрационного удостоверения лекарственного препарата и производитель не являются одной организацией, должны проводиться соответствующие мероприятия, учитывающие принципы настоящей главы.
Заказчик
7.4. Фармацевтическая система качества заказчика должна включать в себя контроль и проверку любой деятельности, переданной для выполнения другому лицу (аутсорсинг). Заказчик гарантирует наличие процедур, обеспечивающих контроль указанной деятельности. Эти процедуры должны включать в себя принципы управления рисками для качества и учитывать нижеприведенные положения.
7.5. До передачи деятельности для выполнения исполнителю заказчик должен убедиться в правомочности, пригодности и компетентности исполнителя в отношении успешного выполнения соответствующих работ. Заказчик также отвечает за включение в соглашение положений, обеспечивающих выполнение требований настоящих Правил.
7.6. Заказчик должен представить исполнителю всю информацию и сведения, необходимые для правильного выполнения предусмотренных в соглашении работ в соответствии с законодательством государств-членов, международными договорами и актами, составляющими право Союза, и регистрационным досье лекарственного препарата. Заказчик должен гарантировать, что исполнитель полностью осведомлен обо всех связанных с продукцией или работой проблемах, которые могут представлять опасность для его помещений, оборудования, персонала, других материалов или продукции.
7.7. Заказчик должен контролировать и проверять действия исполнителя, включая внедрение исполнителем любого необходимого улучшения.
7.8. Заказчик является ответственным за проверку и оценку записей и результатов, связанных с деятельностью, переданной для выполнения другому лицу (аутсорсинг). Заказчик должен убедиться самостоятельно или на основании подтверждения уполномоченного лица исполнителя, что вся продукция и материалы, представленные ему исполнителем, были произведены в соответствии с настоящими Правилами и регистрационным досье лекарственного препарата.
Исполнитель
7.9. Исполнитель должен иметь необходимые помещения, оборудование, а также знания, опыт и компетентный персонал для надлежащего выполнения работ, порученных ему заказчиком.
7.10. Исполнитель должен удостовериться, что все представленные ему продукция, исходные материалы и сведения пригодны для использования по назначению.
7.11. Исполнитель не должен передавать третьей стороне работы или услуги, порученные ему по соглашению, без предварительного рассмотрения и согласования с заказчиком. При заключении соглашения между исполнителем и третьей стороной должна быть обеспечена гарантия того, что информация, включая сведения об оценке соответствия третьей стороны, представляется таким же образом, как между первоначальными заказчиком и исполнителем.
7.12. Исполнитель не должен производить несанкционированные изменения, выходящие за рамки соглашения, поскольку это может неблагоприятно повлиять на качество работ, проводимых для заказчика.
7.13. Исполнитель должен понимать, что работы, передаваемые для выполнения другому лицу (аутсорсинг), включая проведение анализа по контракту, подлежат проверке уполномоченными органами государств-членов.
Соглашение
7.14. Между заказчиком и исполнителем должно быть составлено соглашение, в котором следует определить их взаимные обязательства и процедуры передачи информации, связанные с деятельностью, передаваемой для выполнения другому лицу (аутсорсинга). Технические аспекты соглашения должны составляться компетентными лицами, имеющими соответствующие знания, связанные с указанной деятельностью, согласно настоящим Правилам. Все соглашения о деятельности, передаваемой на аутсорсинг, должны соответствовать законодательству государств-членов и международным договорам и актам, составляющим право Союза, регистрационному досье лекарственного препарата и быть согласованы обеими сторонами.
7.15. В соглашении должно быть однозначно указано, кто отвечает за каждый этап деятельности, передаваемой для выполнения другому лицу (например, управление знаниями, перенос технологии, обеспечение цепи поставки, заключение соглашения с третьей стороной, закупка исходного сырья, материалов и их качество, проведение испытаний и выдача разрешения на использование исходных и упаковочных материалов, проведение производства и контроля качества (включая контроль в процессе производства, отбор образцов и их анализ)).
7.16. Все записи, связанные с деятельностью, передаваемой для выполнения другому лицу, например, записи производства, анализа и реализации продукции, а также соответствующие контрольные образцы должны храниться у заказчика или должны быть ему доступны. Любые записи, относящиеся к оценке качества продукции, в случае предъявления претензий, предполагаемого несоответствия требованиям или при расследовании в случае предположения о фальсификации продукции должны быть доступны заказчику и точно определены в его соответствующих процедурах.
7.17. В соглашении должно быть предусмотрено право заказчика на аудит деятельности, которая выполняется исполнителем или взаимно согласованной третьей стороной.
Глава 8. Претензии, дефекты качества и отзывы продукции
Принцип
В целях защиты здоровья людей и животных должны быть в наличии системы и соответствующие процедуры по регистрации, оценке, расследованию и рассмотрению претензий, в том числе потенциальных дефектов качества, и (при необходимости) по эффективному и оперативному отзыву из сети распределения лекарственных средств для человека или ветеринарных лекарственных средств и исследуемых лекарственных средств. Принципы управления рисками для качества должны применяться при расследовании и оценке дефектов качества, а также в процессе принятия решений в отношении отзыва продукции, корректирующих и предупреждающих действий и других мер по снижению риска. Руководство в отношении этих принципов содержится в главе 1настоящих Правил.
Все заинтересованные уполномоченные органы (организации) государств-членов должны быть своевременно проинформированы в случае подтвержденного дефекта качества лекарственного средства или исследуемого лекарственного средства (производственного дефекта, порчи продукции, выявления фальсификации, несоответствия регистрационному досье или файлу спецификаций продукта или любых других серьезных проблем с качеством), который может привести к отзыву продукции или необычному сокращению поставок. В ситуациях, если продукт на рынке признается не соответствующим регистрационному досье лекарственного препарата, не требуется уведомлять заинтересованные уполномоченные органы (организации) государств-членов при условии, что степень несоответствия удовлетворяет ограничениям, указанным в приложении № 16 к настоящим Правилам, в отношении обращения с незапланированными отклонениями.
В случае деятельности, передаваемой для выполнения другому лицу (аутсорсинга), в соглашении должны быть описаны роли и обязанности производителя, владельца регистрационного досье и (или) спонсора и третьих сторон в отношении оценки, принятия решений, распространения информации и реализации действий по снижению потенциальной опасности, связанной с дефектной продукцией. Руководство в отношении деятельности, передаваемой для выполнения другому лицу (аутсорсинга), приведено в главе 7 настоящих Правил. Такие соглашения также должны содержать контактные данные ответственных лиц каждой стороны для осуществления связи с целью управления дефектами качества и вопросами, связанными с отзывом.
Персонал и организация
8.1. Ответственность за управление расследованием претензий и дефектов качества и принятие решений в отношении мер, которые должны быть приняты для управления любым потенциальным риском, включая отзывы, должен нести соответственно обученный и опытный персонал. Эти лица должны быть независимы от подразделений сбыта и маркетинга организации, если иное не обосновано. Если одним из этих лиц не является уполномоченное лицо, участвующее в сертификации для выпуска соответствующей серии (серий), уполномоченное лицо должно быть своевременно официально проинформировано о любых расследованиях, действиях по снижению риска и любых операциях по отзыву.
8.2. Необходимо иметь достаточное количество обученного персонала и ресурсов для обработки, оценки, расследования и рассмотрения претензий и дефектов качества, для реализации любых действий по снижению риска, а также для управления взаимодействием с уполномоченными органами или организациями государств-членов.
8.3. Должно предусматриваться использование специалистов различных подразделений, включая надлежащим образом обученный персонал по управлению качеством.
8.4. Если работа с претензиями и дефектами качества в организации управляется централизованно, распределение ролей и обязанностей заинтересованных сторон должно быть документировано. При этом централизованное управление не должно приводить к задержкам в расследовании претензий и дефектов качества и управлении ими.
Процедуры обработки и расследования претензий, включая
возможные дефекты качества
8.5. Должны быть в наличии письменные процедуры, описывающие действия, которые необходимо принять при получении претензии. Все претензии должны быть задокументированы и оценены, чтобы установить, представляют ли они собой потенциальный дефект качества или другую проблему.
8.6. Особое внимание следует уделять установлению того, относится ли претензия или подозреваемый дефект качества к фальсификации.
8.7. Поскольку не все претензии, полученные компанией, могут представлять фактические дефекты качества, претензии, которые не свидетельствуют о потенциальном дефекте качества, следует надлежащим образом задокументировать и довести до сведения соответствующей службы или лица, ответственных за расследование и управление претензиями подобного рода, такими как подозреваемые нежелательные явления.
8.8. Должны быть в наличии письменные процедуры, чтобы облегчить запросы о расследовании качества серии лекарственного средства в целях содействия расследованию сообщений о подозреваемых побочных реакциях.
8.9. Если инициируется расследование дефекта качества, должны быть в наличии процедуры, предусматривающие следующее:
i. описание сообщенного дефекта качества;
ii. определение значимости дефекта качества. В рамках этого следует предусматривать проверку или испытания арбитражных и (или) архивных образцов, и в некоторых случаях должен выполняться обзор записей производства серии, записей о сертификации серии и записей о распределении серии (особенно для чувствительной к температуре продукции);
iii. необходимость запросить образец или возвратить всю дефектную продукцию от заявителя и, если образец получен, необходимость выполнения соответствующей оценки;
iv. оценка риска, который представляет дефект качества в зависимости от серьезности и значимости этого дефекта;
v. процесс принятия решений в отношении потенциальной необходимости мер по снижению риска, которые необходимо принять в сети распределения (отзыв серии или продукта или другие действия);
vi. оценка влияния, которое может оказать любое действие по отзыву на доступность лекарственного средства для пациентов (животных) на любом рынке, где обращается данное лекарственное средство, а также необходимость уведомить соответствующие уполномоченные органы государств-членов о таком влиянии;
vii. внутренний и внешний обмен информацией, который должен осуществляться в отношении дефекта качества и его расследования;
viii. идентификация потенциальной причины дефекта качества;
ix. необходимость в определении и осуществлении соответствующих корректирующих и предупреждающих действий, а также в оценке их результативности.
Расследование и принятие решений
8.10. Информация, свидетельствующая о возможных дефектах качества, должна быть зарегистрирована со всеми исходными подробностями. Обоснованность и значимость всех зарегистрированных дефектов качества должны быть документированы и оценены в соответствии с принципами управления рисками для качества с целью обоснования решений, принятых в отношении объема проводимого расследования и предпринимаемых действий.
8.11. Если дефект качества обнаружен или подозревается в какой-то одной серии, должно быть уделено внимание проверке других серий и в некоторых случаях других продуктов, чтобы определить, не оказались ли они также затронуты. В частности, должны быть исследованы другие серии, которые могут содержать части дефектной серии или дефектных компонентов.
8.12. Расследование дефекта качества должно включать обзор предыдущих отчетов о дефектах качества или любую другую соответствующую информацию о любых признаках специфических или повторяющихся проблем, требующих внимания и, возможно, дальнейших регуляторных действий.
8.13. Решения, принятые во время и после расследования дефектов качества, должны отражать уровень риска, который представляет дефект качества, а также серьезность любого несоответствия регистрационному досье (досье исследуемого продукта) или требованиям надлежащей производственной практики. Такие решения должны быть своевременными, чтобы обеспечить поддержание безопасности пациентов и животных соразмерно уровню риска, который может представлять эта проблема.
8.14. Хотя на ранних стадиях расследования полная информация о характере и значимости дефекта качества не всегда может быть доступна, процессы принятия решений все же должны обеспечивать, чтобы соответствующие действия по снижению риска принимались в соответствующих временных точках в ходе таких расследований. Все принятые решения и меры в отношении дефекта качества следует задокументировать.
8.15. В случаях, если дефект качества может привести к отзыву продукции или необычному сокращению поставок продукции, производитель должен своевременно проинформировать об этом держателя регистрационного удостоверения (спонсора) и все заинтересованные уполномоченные органы (организации) государств- членов.
Анализ основных причин, корректирующие и предупреждающие
действия
8.16. В ходе расследования дефектов качества должен применяться соответствующий уровень работы по анализу основных причин. В случаях, если истинная причина дефекта качества не может быть определена, должно быть уделено внимание идентификации наиболее вероятной причины и обращению с ней.
8.17. Если в качестве причины дефекта качества подозревается или определена человеческая ошибка, это должно быть официально обосновано и тщательно рассмотрено, для того чтобы не были упущены из виду возможные процессные, процедурные или системные ошибки или проблемы.
8.18. В отношении дефекта качества должны быть определены и приняты соответствующие корректирующие и предупреждающие действия. Результативность таких действий следует проверить и оценить.
8.19. Записи о дефектах качества следует регулярно просматривать и анализировать тенденции для выявления любых специфических или повторяющихся проблем, требующих внимания.
Отзыв продукции и другие действия по снижению
потенциального риска
8.20. Должны быть установлены письменные процедуры, регулярно пересматриваемые и обновляемые по мере необходимости, в целях осуществления любой деятельности по отзыву или осуществлению любых других действий по снижению риска.
8.21. После размещения продукции на рынке любой возврат ее из сети распределения из-за дефекта качества должен рассматриваться и управляться как отзыв. Это положение не применяется к изъятию (возврату) образцов продукции из сети распределения для содействия расследованию отчетности по дефектам качества.
8.22. Должна быть возможность инициировать операции по отзыву оперативно и в любое время. В некоторых случаях в целях защиты здоровья населения или животных, может потребоваться начать операции по отзыву до установления истинной причины и значимости дефекта качества.
8.23. Записи о дистрибьюции серии (продукции) должны быть легко доступны для лиц, ответственных за отзыв, и содержать достаточную информацию об оптовых покупателях и заказчиках, непосредственно получивших продукцию (с указанием адреса, номеров телефона и (или) факса, работающих круглосуточно, номера серии и количества поставленной продукции), в том числе в отношении экспортируемой продукции и медицинских образцов.
8.24. В отношении исследуемых лекарственных средств должны быть идентифицированы все исследовательские площадки и указаны страны назначения. В отношении исследуемого лекарственного средства, на которое было выдано регистрационное удостоверение, производитель исследуемого лекарственного средства в сотрудничестве со спонсором должен информировать держателя регистрационного удостоверения о любом дефекте качества, который может быть связан с зарегистрированным лекарственным средством. Спонсор должен внедрить процедуру быстрого раскодирования ослепленного продукта, где это необходимо для оперативного отзыва. Спонсор должен обеспечить, чтобы процедура раскрывала идентичность ослепленного продукта только в той мере, которая необходима.
8.25. Следует уделить внимание последующим консультациям с заинтересованными уполномоченными органами или организациями государств-членов по вопросу, насколько далеко должны распространяться действия по отзыву в сети распределения, учитывая потенциальный риск для здоровья населения или животных и любого воздействия, которое могут оказать предлагаемые действия по отзыву. Уполномоченные органы (организации) государств-членов также должны быть проинформированы в ситуациях, когда не ведется никаких действий по отзыву, предложенных для дефектной серии, в связи с истечением срока годности (например, для продукции с коротким сроком годности).
8.26. В случае намерения отозвать продукцию об этом должны быть заранее проинформированы все заинтересованные уполномоченные органы (организации) государств-членов. Для очень серьезных проблем (то есть с возможным серьезным влиянием на здоровье пациентов или животных), можно принимать срочные действия по снижению риска (например, отзыв продукции), предварительно уведомив об этом уполномоченные органы (организации) государств-членов. Везде, где возможно, необходимо стремиться к тому, чтобы такие действия были заблаговременно, до их выполнения, согласованы с заинтересованными уполномоченные органы (организациями) государств-членов.
8.27. Следует также учитывать, могут ли предлагаемые действия по отзыву по-разному повлиять на различные рынки, и, если это так, должны быть разработаны и обсуждены с заинтересованными уполномоченными органами (организациями) государств-членов соответствующие действия по снижению риска для конкретного рынка. До принятия решения о таких мерах по снижению риска, как отзыв, должен быть рассмотрен риск нехватки лекарственного средства, не имеющего зарегистрированной альтернативы, с учетом его терапевтического назначения. Любые решения о непринятии ожидаемых мер по снижению риска должны быть предварительно согласованы с уполномоченными органами (организациями) государства-члена.
8.28. Отозванная продукция должна быть идентифицирована и храниться отдельно в надежном месте до принятия решения о том, как с ней поступить. Должно быть документально оформлено официальное распоряжение в отношении всех отозванных серий продукции. Основание для принятия любого решения о переработке отозванной продукции должно быть задокументировано и обсуждено с соответствующим уполномоченным органом (организацией) государства-члена. Также должен быть учтен остаточный срок годности любых переработанных серий, которые предполагается разместить на рынке.
8.29. Ход процесса отзыва должен регистрироваться до момента завершения и выпуска окончательного отчета, включающего в себя баланс между количеством поставленной и возвращенной продукции (серии продукции).
8.30. Эффективность мероприятий по отзыву следует регулярно оценивать для подтверждения того, что они остаются надежными и пригодными для использования. Такие оценки должны распространяться на ситуации, возникающие как в рабочее, так и в нерабочее время. При выполнении таких оценок следует рассмотреть необходимость выполнения имитации действий по отзыву. Эта оценка должна быть документально оформлена и обоснована.
8.31. В дополнение к отзыву существуют и другие возможные действия по снижению риска, которые могут предприниматься в целях управления рисками, создаваемыми дефектами качества. Такие действия могут включать в себя выпуск предупредительных сообщений для медицинских и ветеринарных работников в отношении использования ими потенциально дефектной серии продукции. Их следует рассматривать в индивидуальном порядке и обсуждать с заинтересованными уполномоченными органами (организациями) государств-членов.
Глава 9. Самоинспекция Принцип
Самоинспекция должна проводиться с целью проверки выполнения предприятием требований настоящих Правил и предложения необходимых корректирующих действий.
9.1. Следует регулярно анализировать вопросы, касающиеся персонала, помещений, оборудования, документации, технологического процесса, контроля качества, реализации лекарственных препаратов, мероприятий по работе с претензиями и в отношении отзывов продукции и деятельности по проведению самоинспекций в соответствии с заранее утвержденной программой в соответствии с определенным графиком проверки их соответствия принципам обеспечения качества.
9.2. Самоинспекция должна проводиться независимо и тщательно специально назначенным квалифицированным лицом (лицами), состоящим в штате предприятия. При необходимости может быть проведен независимый аудит экспертами сторонних организаций.
9.3. Результаты самоинспекций должны быть оформлены документально. Отчеты должны содержать в себе все наблюдения, сделанные в ходе проверки, и, где применимо, предложения по корректирующим действиям. Действия, предпринимаемые по результатам проведенных самоинспекций, также следует оформлять документально.
II. Основные требования к активным фармацевтическим
субстанциям, используемым в качестве исходных материалов
1. Введение
Держатели регистрационных удостоверений и производители лекарственных препаратов должны использовать в качестве исходных материалов только те активные фармацевтические субстанции (далее - АФС), которые произведены с соблюдением настоящих Правил. Эти принципы производства активных фармацевтических субстанций детально изложены в настоящей части Правил.
1.1. Цель
Настоящая часть Правил представляет собой руководящие указания, касающиеся надлежащего производства активных фармацевтических субстанций с соответствующей системой управления качеством. Она также предназначена для помощи в обеспечении качества и чистоты активных фармацевтических субстанций в соответствии с предъявляемыми к ним требованиями.
В настоящей части Правил понятие «производство» включает в себя все виды операций с активными фармацевтическими субстанциями: приемку материалов, производство, упаковку, переупаковку, маркировку, перемаркировку, контроль качества, выдачу разрешения на выпуск, хранение и реализацию, а также соответствующие меры контроля. Понятия «должен», «следует», применяемые в настоящих Правилах, указывают на рекомендации, выполнение которых предполагается, за исключением случаев, когда их выполнить невозможно, или если в соответствующих приложениях не указаны иные требования, или они могут быть заменены альтернативными действиями с эквивалентным уровнем обеспечения качества продукции.
Настоящие Правила в целом не распространяются на вопросы охраны труда персонала, занятого в производстве, а также не затрагивают вопросы защиты окружающей среды. Контроль, осуществляемый в этом случае, является непосредственной обязанностью производителя и регламентируется законодательством государств-членов.
Настоящие Правила не устанавливают требования, предъявляемые при включении активных фармацевтических субстанций в государственный реестр, и не заменяют фармакопейных требований. Они не затрагивают функции уполномоченных органов государств- членов устанавливать особенные требования к включению в государственный реестр лекарственных средств (к получению разрешения (лицензии) на производство) активных фармацевтических субстанций.
1.2. Область применения
Настоящая часть Правил устанавливает требования к производству активных фармацевтических субстанций, используемых в производстве лекарственных препаратов для медицинского и ветеринарного применения. К производству стерильных активных фармацевтических субстанций она применима только до стадии стерилизации и не распространяется на процессы стерилизации и производства стерильных активных фармацевтических субстанций в асептических условиях. Эти процессы следует проводить в соответствии с принципами, предусмотренными настоящими Правилами, и требованиями, предусмотренными приложением № 1 к настоящим Правилам, актами, входящими в право Союза, и законодательством государств-членов.
В случае производства ветеринарных лекарственных средств против эктопаразитов для обеспечения качества могут использоваться другие нормативные документы.
Настоящая часть Правил не распространяется на цельную донорскую кровь и плазму, поскольку требования по забору и испытанию крови регулируются соответствующими нормативными правовыми актами государств-членов, однако распространяется на активные фармацевтические субстанции, получаемые с использованием донорской крови или плазмы в качестве исходного сырья. Настоящая часть Правил не распространяется на нерасфасованные лекарственные средства и применяется ко всем другим активным исходным материалам с учетом любых отступлений, предусмотренных приложениями к настоящим Правилам, в частности приложениями № 2 - 7, где указаны дополнительные требования к активным фармацевтическим субстанциям.
Раздел 19 настоящей части Правил содержит требования, распространяющиеся только на производство активных фармацевтических субстанций, используемых для получения лекарственных препаратов, предназначенных для клинических исследований.
Под исходными материалами для производства активных фармацевтических субстанций понимаются сырье, промежуточные продукты или другие активные фармацевтические субстанции, которые используются в производстве активных фармацевтических субстанций и которые как важный структурный фрагмент вводятся в структуру активной фармацевтической субстанции. Исходные материалы для производства активных фармацевтических субстанций могут быть закуплены по соглашению у одного или нескольких поставщиков либо производиться самостоятельно. Исходные материалы для производства активных фармацевтических субстанций, как правило, имеют установленные химические свойства и структуру.
Производитель должен определить и документально обосновать стадию, с которой должно начинаться производство активной фармацевтической субстанции. Для процессов синтеза эта стадия определяется как стадия ввода в технологический процесс исходных материалов для производства активных фармацевтических субстанций. Для других процессов (ферментации, экстракции, очистки и пр.) данную стадию определяют с учетом конкретных особенностей производства. В таблице «Применение положений части II настоящих Правил к производству АФС» приведены руководящие указания относительно момента, когда обычно вводят в процесс исходные материалы для производства активных фармацевтических субстанций. Начиная с этой стадии, на данные промежуточные продукты и (или) стадии производства активных фармацевтических субстанций действуют требования настоящей части Правил. Они включают в себя валидацию критических стадий производственного процесса, оказывающих влияние на качество активных фармацевтических субстанций. В то же время выбор производителем стадии технологического процесса для проведения валидации не обязательно означает, что эта стадия является критической. Требования настоящей части Правил распространяются, как правило, на стадии, выделенные в указанной таблице серым цветом. Это не означает, что в процессе производства должны выполняться все стадии, указанные в данной таблице. Строгость следования требованиям настоящей части Правил должна возрастать от ранних стадий производства активных фармацевтических субстанций к завершающим стадиям технологического процесса, очистки и упаковки. Такую обработку физическими методами активных фармацевтических субстанций, как гранулирование, покрытие оболочкой или физическое изменение размера частиц (например, грубый и тонкий помол), следует проводить в соответствии с требованиями настоящих Правил. Настоящая часть Правил не применяется к стадиям, которые предшествуют введению в процесс веществ, определенных как исходные материалы для производства активных фармацевтических субстанций.
В части II Правил используется понятие «активная фармацевтическая субстанция», которое следует рассматривать как синоним понятия «активный фармацевтический ингредиент» (АФИ). Понятия, употребляемые в настоящей части Правил, и их определения (которые приведены в разделе 20 «Термины и определения») следует применять только в части II настоящих Правил. Для аналогичных понятий, употребляемых в части I настоящих Правил, приведены определения в общем разделе «Термины и определения» настоящих Правил, следовательно, их следует применять только в контексте части I настоящих Правил.
Таблица 1
|
Тип производства |
Наименование стадии производства АФС, на которые распространяется действие положений части II настоящих Правил (выделены серым цветом) |
||||
|
Химическое производство |
производство исходного сырья для АФС |
введение в процесс исходного сырья для производства АФС |
производство промежуточного продукта |
выделение и очистка |
обработка физическими методами и упаковка |
|
АФС, получаемые из сырья животного происхождения |
сбор органов, жидкостей или тканей |
измельчение, смешивание и (или) первичная обработка |
введение в процесс исходного сырья для производства АФС |
выделение и очистка |
обработка физическими методами и упаковка |
|
АФС, получаемые из сырья растительного происхождения |
сбор растений |
измельчение и первичная экстракция |
введение в процесс исходного сырья для производства АФС |
выделение и очистка |
обработка физическими методами и упаковка |
|
Растительные экстракты, используемые в качестве АФС |
сбор растений |
измельчение и первичная экстракция |
- |
дальнейшая экстракция |
обработка физическими методами и упаковка |
|
АФС, состоящие из размельченных или растертых в порошок растений |
сбор растений и (или) культивирование и сбор |
измельчение (растирание, дробление) |
- |
- |
обработка физическими методами и упаковка |
|
Биотехнология: ферментация (культивирование клеток) |
создание главного и рабочего банков клеток |
поддержание рабочего банка клеток |
культивирование клеток и (или) ферментация |
выделение и очистка |
обработка физическими методами и упаковка |
|
«Классическая» ферментация для производства АФС |
создание банка клеток |
поддержание банка клеток |
ввод клеток в процесс ферментации |
выделение и очистка |
обработка физическими методами и упаковка |
2. Управление качеством
2.1. Принципы
2.10. Ответственность за качество должен нести весь персонал, занятый в производстве.
2.11. Каждый производитель должен разработать, документально оформить и внедрить эффективную систему управления качеством при активном участии руководящего и соответствующего производственного персонала.
2.12. Система управления качеством должна охватывать организационную структуру, процедуры, процессы и ресурсы, а также деятельность, необходимую для обеспечения гарантии соответствия АФС всем требованиям соответствующих спецификаций в отношении качества и чистоты. Следует определить и оформить документально все виды деятельности, имеющие отношение к качеству.
2.13. Необходимо иметь независимый от производственного отдела отдел (отделы) качества, который выполняет функции обеспечения качества и контроля качества. Это могут быть либо отдельные службы обеспечения качества и контроля качества, либо одно лицо или группа лиц в зависимости от размеров и структуры организации.
2.14. Необходимо точно определить лиц, уполномоченных выдавать разрешение на выпуск промежуточной продукции и АФС.
2.15. Все действия, имеющие отношение к качеству, следует оформлять документально непосредственно при их выполнении.
2.16. Любое отклонение от установленных процедур следует оформлять документально и обосновывать. Необходимо проводить расследование критических отклонений, а также оформлять документально это расследование и сделанные выводы.
2.17. Материалы не могут быть разрешены к выпуску или использованы до получения удовлетворительного заключения по результатам оценки, проведенной отделом (отделами) качества, если на предприятии не существует соответствующих систем, разрешающих такое использование (например, выпуск в статусе карантина, как описано в пункте 10.20 настоящей части Правил, либо использование сырья или промежуточной продукции, оценка качества которых еще не завершена).
2.18. Следует разработать процедуры своевременного оповещения ответственных руководящих лиц о результатах инспекций уполномоченных органов, серьезных недостатках в отношении соблюдения требований настоящих Правил, дефектах продукции и о принятии соответствующих мер (например, о претензиях в отношении качества, отзывах, действиях уполномоченных органов и др.).
2.19. Для достижения цели управления качеством должна быть внедрена всесторонне разработанная и правильно функционирующая система качества, включающая в себя надлежащую производственную практику, контроль качества и управление рисками для качества.
2.2. Управление рисками для качества
2.20. Управление рисками для качества является систематическим процессом оценки, контроля, передачи информации и обзора рисков для качества АФС. Этот процесс можно осуществлять как перспективно, так и ретроспективно.
2.21. Система управления рисками для качества должна гарантировать, что:
оценка рисков базируется на научных знаниях, опыте производства и в конечном счете связана с защитой пациента путем обмена информацией с потребителем АФС;
уровень усилий, формализации и документального оформления процесса управления рисками для качества соизмерим с уровнем рисков.
Примеры процессов и применения управления рисками для качества приведены в части III настоящих Правил.
2.3. Обязанности отдела (отделов) качества
2.30. Отдел качества должен быть вовлечен в решение всех вопросов, относящихся к качеству.
2.31. Отдел качества должен рассматривать и согласовывать все документы, имеющие отношение к качеству продукции.
2.32. Основные обязанности независимого отдела качества не подлежат передаче другим службам. Эти обязанности должны быть представлены в письменном виде и включать следующее (но не обязательно ограничиваться этим):
выдача разрешения на выпуск или отклонение всех АФС. Выдача разрешения на выпуск или отклонение промежуточной продукции, предназначенной для использования вне сферы контроля предприятия- производителя;
создание системы выдачи разрешения на выпуск или отклонения исходного сырья, промежуточной продукции, материалов для упаковки и маркировки;
проверка заполненных записей по производству серии и документов лабораторного контроля в отношении критических стадий процесса перед выдачей разрешения на выпуск АФС для реализации;
обеспечение расследования причин критических отклонений и их устранение;
согласование или утверждение всех спецификаций и основных производственных инструкций;
согласование или утверждение всех процедур, которые могут оказывать влияние на качество промежуточной продукции или АФС;
обеспечение проведения внутренних аудитов (самоинспекций); одобрение производителей промежуточной продукции и АФС, работающих по контракту;
утверждение изменений, которые потенциально могут повлиять на качество промежуточной продукции или АФС;
рассмотрение и утверждение протоколов и отчетов по валидации; обеспечение проведения расследования и принятия решений по претензиям, связанным с качеством;
подтверждение того, что системы, использующиеся для технического обслуживания, калибровки и поверки критического оборудования, являются эффективными;
обеспечение испытаний исходного сырья и материалов и документального оформления результатов;
обеспечение наличия данных о стабильности для подтверждения устанавливаемых дат проведения повторных испытаний или истечения сроков годности, а также условий хранения АФС и (или) промежуточной продукции в тех случаях, когда это целесообразно;
проведение обзоров качества продукции (согласно указаниям, приведенным в пункте 2.6 настоящей части Правил).
2.4. Обязанности по производственной деятельности
Обязанности по производственной деятельности должны быть представлены в письменном виде и включать следующее (но не обязательно ограничиваться этим):
разработка, пересмотр, утверждение и распределение инструкций по производству промежуточной продукции или АФС в соответствии с утвержденной процедурой;
производство АФС и, при необходимости, промежуточной продукции в соответствии с заранее утвержденными инструкциями;
рассмотрение всех записей по производству серии продукции и подтверждение их заполнения и подписания;
обеспечение обязательного документирования всех отклонений от процесса производства и проведения их оценки, а также расследования всех критических отклонений и документального оформления полученных выводов;
обеспечение чистоты производственных помещений и, при необходимости, их дезинфекции;
обеспечение выполнения необходимых калибровок, а также ведения и хранения записей;
обеспечение обслуживания помещений и оборудования, а также ведения и хранения записей;
обеспечение проверки и согласования протоколов валидации и отчетов;
оценка предлагаемых изменений в отношении продукции, процесса или оборудования;
обеспечение квалификации новых и, при необходимости, модернизированных помещений и оборудования.
2.5. Внутренние аудиты (самоинспекция)
2.50. Для подтверждения соответствия производства АФС требованиям настоящих Правил следует регулярно проводить внутренние аудиты согласно утвержденному графику.
2.51. Результаты внутреннего аудита и последующие корректирующие действия следует оформлять документально и доводить до сведения ответственных руководителей предприятия. Согласованные корректирующие действия следует выполнять своевременно и эффективно.
2.6. Обзор качества продукции
2.60. Для подтверждения постоянства процесса следует регулярно проводить обзор качества АФС. Такие обзоры качества следует проводить, как правило, ежегодно с последующим документальным оформлением. Они должны включать в себя, по крайней мере, следующее:
обзор результатов контроля в процессе производства по критическим точкам и испытаний АФС по критическим показателям;
обзор всех серий, не соответствующих утвержденным спецификациям;
обзор всех критических отклонений или несоответствий и связанных с ними расследований;
обзор любых изменений, внесенных в процессы или аналитические методики;
обзор результатов программы изучения стабильности;
обзор всех возвратов, претензий и отзывов, связанных с качеством;
обзор адекватности корректирующих действий.
2.61. Необходимо анализировать результаты этого обзора и оценивать, следует ли предпринимать корректирующее действие или проводить повторную валидацию. Обоснование необходимости такого корректирующего действия должно быть оформлено документально. Согласованные корректирующие действия следует осуществлять своевременно и эффективно.
3. Персонал
3.1. Квалификация персонала
3.10. Должно быть достаточное количество персонала, имеющего соответствующее образование, подготовку и(или) практический опыт для осуществления производства промежуточной продукции и АФС, а также надзора за их производством.
3.11. Обязанности всего персонала, занятого в производстве промежуточной продукции и АФС, должны быть точно определены и изложены в письменной форме.
3.12. Необходимо регулярно проводить обучение персонала с привлечением квалифицированных специалистов, как минимум, по вопросам, связанным с конкретными операциями, выполняемыми сотрудником, а также с требованиями настоящих Правил, имеющими отношение к функциональным обязанностям сотрудника. Следует вести записи обучения, а само обучение следует периодически оценивать.
3.2. Гигиена персонала
3.20. Персонал должен соблюдать правила гигиены.
3.21. Персонал должен носить чистую, соответствующую его производственной деятельности одежду, которую при необходимости следует менять. Чтобы защитить промежуточную продукцию и АФС от контаминации, при необходимости следует использовать дополнительную защитную одежду, закрывающую голову, лицо, руки и кисти рук.
3.22. Персонал должен избегать непосредственного контакта с промежуточной продукцией или АФС.
3.23. Курение, жевание, прием пищи, питье и хранение пищевых продуктов допускаются только в специально предназначенных зонах, отделенных от производственных зон.
3.24. При наличии у сотрудников инфекционных заболеваний или открытых повреждений на незащищенных участках поверхности тела их следует отстранить от участия в работе, поскольку при этом качество АФС подвергается риску. Любого сотрудника с явными признаками заболевания или открытыми повреждениями кожи (по результатам медицинского обследования или наблюдения) следует отстранить от работ, при выполнении которых состояние его здоровья может оказать неблагоприятное воздействие на качество АФС, до выздоровления или получения медицинского заключения о том, что сотрудник допущен к работе.
3.3. Консультанты
3.30. Консультанты по вопросам производства и контроля промежуточной продукции или АФС должны иметь соответствующее образование, подготовку, практический опыт или любое их сочетание для того, чтобы консультировать по вопросам, для решения которых их пригласили.
3.31. Следует вести их учет с указанием личных данных, адреса проживания, квалификации и вида услуг, предоставляемых этими консультантами.
4. Здания и помещения
4.1. Проектирование и строительство
4.10. Здания и помещения, используемые при производстве промежуточной продукции и АФС, следует располагать, проектировать и конструировать таким образом, чтобы обеспечить возможность их очистки, обслуживания и функционирования в соответствии с типом и стадией производства. Помещения следует проектировать таким образом, чтобы свести к минимуму возможную контаминацию. Если установлены спецификации в отношении микробиологических показателей промежуточной продукции или АФС, помещения следует проектировать таким образом, чтобы по возможности ограничить риск нежелательной микробной контаминации.
4.11. Здания и помещения должны быть достаточно просторными для правильного расположения оборудования, хранения и перемещения материалов, чтобы предотвратить перепутывание и контаминацию.
4.12. Если оборудование (например, закрытые или изолированные системы) обеспечивает надежную защиту материалов, оно может быть расположено вне помещения.
4.13. Перемещение материалов и передвижение персонала в здании и помещениях должно быть предусмотрено таким образом, чтобы предотвратить перепутывание или контаминацию.
4.14. Следует определить конкретные зоны или другие системы контроля для следующих операций:
приемка, идентификация, отбор проб и карантин поступающих материалов до выдачи разрешения на использование или до отклонения;
хранение промежуточной продукции и АФС в карантине до выдачи разрешения на выпуск или до отклонения;
отбор проб промежуточной продукции и АФС; хранение отклоненных материалов до избавления от них (например, возврат, повторная обработка или уничтожение);
хранение материалов, которые разрешены к использованию; технологические операции; операции по упаковке и маркировке; проведение лабораторных анализов.
4.15. Следует предусмотреть наличие необходимых помещений для подготовки персонала (мытье рук и др.) в и туалетных комнат в достаточном количестве и поддерживать в них чистоту. К ним должна быть подведена горячая и холодная вода, должно быть в наличии мыло или иное моющее средство, воздушные сушилки или одноразовые полотенца. Умывальники и туалетные комнаты должны быть отделены от производственных зон, но легкодоступны. Следует обеспечить наличие помещений для переодевания и при необходимости для принятия душа.
4.16. Лабораторные зоны (работы) следует, как правило, отделять от производственных зон. Некоторые лабораторные зоны, в частности, зоны, используемые для контроля в процессе производства, можно размещать в производственных зонах при условии, что операции технологического процесса не оказывают неблагоприятного влияния на точность лабораторных измерений, а лабораторные работы не оказывают неблагоприятного воздействия на технологический процесс, промежуточную продукцию или АФС.
4.2. Инженерные системы
4.20. Все инженерные системы, которые могут повлиять на качество продукции (например, пар, газы, сжатый воздух, а также системы нагревания, вентиляции и кондиционирования воздуха), должны пройти квалификацию. Их следует соответствующим образом контролировать и предпринимать меры, если превышены допустимые пределы. Должны иметься в наличии чертежи этих инженерных систем.
4.21. При необходимости следует предусматривать соответствующие системы вентиляции и фильтрации воздуха, а также вытяжные устройства. Эти системы должны быть спроектированы и сконструированы таким образом, чтобы свести к минимуму риск контаминации и перекрестной контаминации. Они также должны быть снабжены оборудованием для контроля давления воздуха, наличия микроорганизмов (если это необходимо), запыленности, влажности и температуры, если это требуется для данной стадии производства. Особое внимание следует уделять зонам, в которых АФС подвергаются воздействию производственной среды.
4.22. В производственных помещениях с рециркуляцией воздуха следует предусмотреть меры по предотвращению риска контаминации и перекрестной контаминации.
4.23. Стационарные трубопроводы следует должным образом идентифицировать. Это можно сделать с помощью маркировки отдельных трубопроводов, соответствующей документации, систем компьютерного контроля или иными способами. Трубопроводы следует располагать таким образом, чтобы избежать риск контаминации промежуточной продукции или АФС.
4.24. Стоки должны иметь соответствующие размеры и обеспечивать разрыв струи или, если это необходимо, иметь устройство для предотвращения обратного потока.
4.3. Вода
4.30. Должно быть подтверждено, что вода, используемая в производстве АФС, соответствует предполагаемому назначению.
4.31. Если нет других указаний, качество воды, используемой в производственном процессе, должно как минимум соответствовать требованиям нормативных документов государств-членов к питьевой воде.
4.32. Если для обеспечения качества АФС характеристик питьевой воды недостаточно и необходимы более жесткие требования к химическим и (или) микробиологическим характеристикам воды, должны быть разработаны соответствующие спецификации на воду по физическим (химическим) свойствам, общему числу микроорганизмов, недопустимым микроорганизмам и (или) содержанию эндотоксинов в воде.
4.33. Если производитель подвергает воду, используемую в производстве, специальной обработке для достижения определенного качества, то процесс обработки должен пройти валидацию и его следует контролировать с учетом установленных пределов.
4.34. Если производитель нестерильной АФС намеревается использовать свою продукцию для последующего производства стерильного лекарственного препарата или утверждает, что его продукция пригодна для получения стерильного лекарственного препарата, то воду, используемую на последних стадиях выделения и очистки, следует подвергать мониторингу и контролировать в отношении общего количества микроорганизмов, недопустимых микроорганизмов и эндотоксинов.
4.4. Разделение зон
4.40. Производство продукции с высокой сенсибилизирующей активностью, такой как пенициллины или цефалоспорины, следует осуществлять в специально выделенных производственных зонах, которые могут включать помещения, оборудование для обработки воздуха и технологическое оборудование.
4.41. Специально выделенные производственные зоны должны быть предусмотрены также для веществ с инфицирующими свойствами, с высокой фармакологической активностью или токсичностью (например, некоторые стероиды или цитотоксические противоопухолевые средства), за исключением тех случаев, когда установлены и осуществляются валидированные процедуры инактивации и (или) очистки.
4.42. Следует разработать и ввести в действие соответствующие меры для предотвращения перекрестной контаминации со стороны персонала, материалов и иных объектов, перемещающихся из одной выделенной зоны в другую.
4.43. Любые технологические операции (включая взвешивание, размол или упаковку) с высокотоксичными нефармацевтическими веществами, такими как гербициды и пестициды, не допускается проводить в тех же зданиях и (или) на том же оборудовании, которые используются для производства АФС. Работу с такими высокотоксичными нефармацевтическими веществами и их хранение следует осуществлять отдельно от АФС.
4.5. Освещение
4.50. Во всех зонах должно быть обеспечено соответствующее освещение для того, чтобы проводить очистку, техническое обслуживание и надлежащее выполнение операций.
4.6. Стоки и отходы
4.60. Стоки, отходы и другие отработанные материалы (например, твердые, жидкие и газообразные побочные продукты производства) внутри и снаружи зданий, а также на непосредственно прилегающей территории следует удалять своевременно, обеспечивая безопасность и соблюдая санитарно-гигиенические нормы. Контейнеры для мусора и (или) сточные трубы должны быть четко промаркированы.
4.7. Санитарная обработка и техническое обслуживание
4.70. Здания, используемые при производстве промежуточной продукции и АФС, следует надлежащим образом обслуживать, ремонтировать и содержать в чистоте.
4.71. Следует разработать письменные процедуры, в которых определена ответственность за проведение санитарной обработки, приведены графики проведения очистки, перечислены методы, оборудование и материалы, используемые при очистке зданий и помещений.
4.72. При необходимости следует также разработать письменные процедуры по использованию соответствующих средств против грызунов, инсектицидов, фунгицидов, фумигантов и средств очистки и дезинфекции, чтобы избежать контаминации оборудования, исходного сырья, упаковочных и маркировочных материалов, промежуточной продукции и АФС.
5. Технологическое оборудование
5.1. Проектирование и монтаж
5.10. Оборудование, используемое при производстве промежуточной продукции и АФС, должно иметь соответствующую конструкцию, соответствующие размеры и располагаться в соответствии со своим назначением и таким образом, чтобы персонал мог осуществлять очистку, дезинфекцию (при необходимости) и техническое обслуживание.
5.11. Поверхности оборудования, контактирующие с сырьем, промежуточными продуктами или АФС, не должны влиять на качество промежуточных продуктов и АФС и приводить к выходу показателей их качества за пределы допустимых значений, установленных в утвержденных спецификациях.
5.12. Технологическое оборудование следует использовать только в пределах рабочих диапазонов, установленных при квалификации.
5.13. Основное оборудование (например, реакторы, емкости для хранения) и стационарные технологические линии, используемые в производстве промежуточной продукции или АФС, следует соответствующим образом идентифицировать.
5.14. Любые вещества, необходимые для функционирования оборудования, такие как смазки, жидкости для нагрева или хладагенты, не должны контактировать с промежуточной продукцией или АФС, чтобы не изменять качество промежуточной продукцией или АФС за пределы допустимых значений, установленных в спецификациях.
Следует провести оценку любых отклонений от этого требования, чтобы убедиться, что отсутствует неблагоприятное воздействие этих материалов, влияющее на их пригодность для использования. При возможности следует использовать смазки и масла, предназначенные для пищевой промышленности.
5.15. Во всех случаях, когда это применимо, следует использовать закрытое или герметично закрывающееся оборудование. При использовании открытого оборудования или его открытии необходимо принять меры предосторожности, чтобы свести к минимуму риск контаминации.
5.16. Должен иметься в наличии актуальный комплект чертежей используемого оборудования, а также критических систем обвязки (например, контрольно-измерительных приборов, вспомогательных систем).
5.2. Техническое обслуживание и очистка оборудования
5.20. Профилактическое техническое обслуживание оборудования следует проводить в соответствии с утвержденными графиками и процедурами, в которых должна быть определена ответственность за их проведение.
5.21. Необходимо разработать письменные инструкции по очистке оборудования и последующего получения разрешения на его использование при производстве промежуточной продукции и АФС. Процедуры очистки должны быть описаны достаточно подробно, чтобы операторы могли проводить очистку оборудования любого типа воспроизводимым и эффективным способом. Эти процедуры должны включать в себя:
установление ответственности за очистку оборудования;
графики очистки, включая (при необходимости) графики санитарной обработки;
полное описание методов и материалов, включая приготовление моющих средств, используемых для очистки оборудования;
инструкции по разборке и сборке каждой (при необходимости) единицы оборудования для обеспечения надлежащей очистки; инструкции по удалению маркировки предыдущей серии; инструкции по защите чистого оборудования от контаминации перед его использованием;
порядок проверки чистоты оборудования непосредственно перед его использованием, если это практически осуществимо;
установление (когда применимо) максимального промежутка времени между окончанием процесса и очисткой оборудования.
5.22. Оборудование и принадлежности следует очищать, хранить и при необходимости подвергать дезинфекции или стерилизовать для предотвращения контаминации или переноса материала, которые могут приводить к выходу показателей качества промежуточной продукции и АФС за пределы, установленные в спецификациях.
5.23. В отношении оборудования, предназначенного для непрерывного технологического процесса или для производства кампаниями последовательных серий одной и той же промежуточной продукции или одной и той же АФС, следует проводить очистку через определенные промежутки времени во избежание накопления и переноса контаминантов (например, продуктов разложения или недопустимого количества микроорганизмов).
5.24. Оборудование, используемое для производства разных материалов, следует очищать в промежутках между их сменой во избежание перекрестной контаминации.
5.25. Следует установить критерии приемлемости в отношении остатков, а также обосновать эти критерии и выбор процедур очистки и моющих средств.
5.26. Оборудование должно быть соответствующим образом промаркировано в отношении его содержимого и состояния чистоты.
5.3. Калибровка
5.30. Калибровку контрольно-измерительного и аналитического оборудования (в том числе весов, приборов для мониторинга), имеющего критическое значение для обеспечения качества промежуточной продукции или АФС, следует проводить в соответствии с письменными инструкциями и установленным графиком.
5.31. Калибровку следует проводить с использованием стандартных образцов, прослеживаемых до соответствующих сертифицированных стандартных образцов (если таковые существуют).
5.32. Записи проведения калибровок должны сохраняться.
5.33. Текущий статус калибровки критического оборудования должен быть известен и доступен для его проверки.
5.34. Не следует использовать приборы, не соответствующие критериям калибровки.
5.35. Отклонения от утвержденных стандартов калибровки для критических приборов необходимо расследовать, чтобы определить, повлияло ли это на качество промежуточной продукции и АФС, произведенных с использованием данного оборудования после его последней успешной калибровки.
5.4. Компьютеризированные системы
5.40. Компьютеризированные системы, относящиеся к надлежащей производственной практике, подлежат валидации. Глубина и масштаб валидации зависят от многообразия, сложности и критичности применения компьютеризированных систем.
5.41. Надлежащие квалификация монтажа и квалификация функционирования должны продемонстрировать пригодность компьютерного оборудования и программного обеспечения для выполнения поставленных задач.
5.42. Поставляемое на рынок программное обеспечение, которое было квалифицировано, не требует проведения испытаний того же уровня. Если существующая система не прошла валидацию во время установки, при наличии соответствующей документации можно провести ретроспективную валидацию.
5.43. Необходимо, чтобы компьютеризированные системы имели достаточный уровень контроля для предотвращения несанкционированного доступа к данным или изменения данных. Необходимо предусмотреть меры по предотвращению недостаточности данных (например, в связи с отсутствием регистрации данных вследствие отключения системы). Должна регистрироваться информация о любых изменениях данных, о последнем вводе данных, о том, кем и когда они были сделаны.
5.44. Необходимо иметь письменные процедуры по эксплуатации и техническому обслуживанию компьютеризированных систем.
5.45. Если критические данные вводятся вручную, следует предусмотреть дополнительную проверку точности их введения. Такую проверку может выполнить второй оператор или сама система.
5.46. Сбои в работе компьютеризированных систем, которые могут повлиять на качество промежуточной продукции или АФС, на достоверность записей или результатов испытаний, следует оформлять документально и расследовать.
5.47. Изменения в компьютеризированных системах необходимо осуществлять в соответствии с процедурами внесения изменений, официально их санкционировать, оформлять документально и тестировать. Следует сохранять записи всех изменений, включая модификацию и усовершенствование компьютерного оборудования, программного обеспечения и других критических компонентов системы. Эти записи должны быть доказательством того, что система поддерживается в валидированном состоянии.
5.48. Если сбой или поломка системы приводят к постоянной потере записей, следует предусмотреть систему резервного копирования информации. Во всех компьютеризированных системах должны быть предусмотрены средства, обеспечивающие защиту данных.
5.49. В дополнение к компьютерной системе допускается запись данных и другим способом.
6. Документация и записи
6.1. Система документации и спецификации
6.10. Все документы, имеющие отношение к производству промежуточной продукции или АФС, следует составлять, проверять, утверждать и распространять в соответствии с письменными процедурами. Документы могут быть оформлены как в письменном виде, так и в электронном виде.
6.11. Выдачу, пересмотр, замену или изъятие всей документации следует контролировать с сохранением сведений об их предыдущих версиях.
6.12. Следует организовать систему хранения всех документов (например, отчетов о разработке, масштабировании, передаче технологий, валидации процесса, записей обучения и производства, документов по контролю и записей реализации). Необходимо указать сроки хранения этих документов.
6.13. Все записи производства, контроля и распределения следует хранить не менее 1 года после истечения срока годности серии. Записи, содержащие данные повторных испытаний АФС, следует сохранять не менее 3 лет после полной реализации серии.
6.14. Записи следует выполнять несмываемыми чернилами в специально предусмотренных для этого местах сразу же после выполнения операций. Лицо, сделавшее запись, должно быть обозначено. Исправления в записях должны быть датированы и подписаны и не должны препятствовать прочтению записи в ее первоначальном виде.
6.15. В течение периода хранения оригиналы записей или их копии должны быть легкодоступны на предприятии, где осуществлялись работы, описанные в этих записях. Если обеспечивается быстрое получение записей с помощью электронных или других средств, допускается использование других мест для их хранения.
6.16. Спецификации, инструкции, процедуры и записи можно хранить либо в оригинале, либо в виде копий (например, фотокопии, микрофильмы, микрофиши или др.). Если использовались методы уменьшения оригинала (например, микрофильмирование, электронные записи), необходимо иметь соответствующее считывающее оборудование, а также средства для изготовления печатных копий.
6.17. Необходимо разработать и оформить документально спецификации на исходное сырье, промежуточную продукцию (при необходимости), АФС и материалы для маркировки и упаковки. Дополнительно могут понадобиться спецификации на некоторые другие материалы, такие как вспомогательные материалы, прокладки или другие материалы, используемые в ходе производства промежуточной продукции или АФС, которые могут быть критическими для качества. Следует установить и оформить документально критерии приемлемости для контроля в процессе производства.
6.18. Если используются электронные подписи на документах, они должны быть идентифицированы и защищены.
6.2. Записи об очистке и использовании оборудования
6.20. В записях об использовании, очистке, санитарной обработке и (или) стерилизации, а также о техническом обслуживании основного оборудования должны быть указаны: дата, время (при необходимости), наименование продукции, номер каждой серии произведенной на этом оборудовании продукции, а также наименование лица, которое проводило очистку и техническое обслуживание.
6.21. Не требуется составление отдельных записей об очистке и использовании оборудования в случае, если оно специально предназначено для производства одного наименования промежуточного продукта или АФС, и серии этого промежуточного продукта или АФС производятся в прослеживаемой последовательности. В случае использования специально предназначенного оборудования записи о его очистке, техническом обслуживании и эксплуатации могут быть частью досье на серию или отдельным документом.
6.3. Записи на исходное сырье, промежуточные продукты,
материалы для маркировки и упаковки АФС
6.30. Необходимо вести записи на исходное сырье, промежуточные продукты, материалы для маркировки и упаковки АФС, содержащие следующие данные:
наименование производителя, идентификация и количество каждой поставки каждой серии исходного сырья, промежуточной продукции или материалов для маркировки или упаковки АФС;
наименование поставщика, контрольный номер (номера) поставщика (при наличии) или другой идентификационный номер, номер, присвоенный при приемке, и дата приемки;
результаты всех проведенных испытаний или проверок и сделанные на их основании выводы;
записи, в которых прослеживается использование исходного сырья и материалов;
документация по оценке и проверке материалов для маркировки и упаковки АФС, подтверждающая соответствие установленным спецификациям;
окончательное решение относительно отклоненного исходного сырья, промежуточной продукции или материалов для маркировки и упаковки АФС.
6.31. Следует хранить утвержденные образцы этикеток для сравнения с ними выпускаемых этикеток.
6.4. Основные технологические инструкции
6.40. Для обеспечения однородности продукта от серии к серии необходимо, разработать основные технологические инструкции для каждого вида промежуточной продукции и АФС, подписанные и датированные одним лицом, а также независимо проверенные подписанные и датированные лицом из отдела качества.
6.41. Основные технологические инструкции должны включать в себя:
наименование выпускаемой промежуточной продукции или АФС и стадии технологического процесса, а также, если применимо, соответствующий код документа;
полный перечень исходного сырья и промежуточной продукции с указанием наименований или кодов, достаточно специфичных для того, чтобы можно было провести идентификацию и определить любые специальные показатели качества;
точное указание количества или соотношение каждого наименования используемого сырья или промежуточной продукции с указанием единиц измерения. Если такое количество не является фиксированным, то необходимо привести расчет для каждого размера серии или режима технологического процесса. Следует привести отклонения от указанных количеств, если они обоснованы;
место осуществления технологического процесса и основное технологическое оборудование, которое при этом используется;
подробные технологические действия, в том числе последовательность, которую необходимо соблюдать, и используемые диапазоны параметров процесса;
указания по отбору проб и контролю в процессе производства с указанием критериев приемлемости (при необходимости);
предельные сроки завершения отдельных стадий технологического процесса и (или) процесса в целом (при необходимости);
ожидаемые диапазоны выхода продукции на соответствующих стадиях процесса или в определенное время;
особые указания и меры предосторожности, которые следует соблюдать, или соответствующие перекрестные ссылки на них (при необходимости);
указания по хранению промежуточной продукции или АФС для обеспечения их пригодности к использованию, включая материалы для маркировки и упаковки, а также особые условия хранения с указанием сроков (при необходимости).
6.5. Записи по производству и контролю качества серии
(досье на серию)
6.50. Для каждого промежуточного продукта и АФС должны быть подготовлены записи по производству и контролю качества, которые могут быть собраны в досье на серию, включающее в себя полную информацию о производстве и контроле качества каждой серии. Внесение записей по производству серии осуществляется по форме, которая должна соответствовать технологической инструкции и являться актуальной версией. Если указанная форма составлена на основании отдельной части технологической инструкции, такой документ должен содержать ссылку на используемую действующую технологическую инструкцию.
6.51. Формы для записей должны быть пронумерованы с указанием конкретного номера серии или идентификационного номера, датированы и подписаны при выдаче. При непрерывном производстве код продукции, а также дата и время выпуска могут служить однозначными идентификаторами до присвоения окончательного номера серии.
6.52. В записях по производству и контролю качества (досье на серию) продукции после завершения каждой важной технологической стадии следует указывать следующие сведения:
дата и время (при необходимости);
основное используемое оборудование (например, реакторы, сушилки, мельницы и др.);
специфическая идентификация каждой серии, включая массу, единицы измерения, номера серий исходного сырья, промежуточной продукции или любых материалов, прошедших повторную обработку в ходе производства;
зарегистрированные фактические результаты критических параметров процесса;
данные о любых проведенных отборах проб;
подписи лиц, выполнявших каждую критическую стадию при работе, а также осуществлявших непосредственный надзор или проверку;
результаты испытаний в процессе производства и лабораторных испытаний;
фактический выход на соответствующих стадиях или в определенное время;
описание упаковки и маркировки для промежуточной продукции или АФС;
образец этикетки для АФС или промежуточной продукции, если они произведены для продажи;
любое замеченное отклонение, его оценка, информация о проведенном расследовании (при необходимости) или ссылка на такое расследование, если соответствующие документы хранятся отдельно;
результаты контроля при выдаче разрешения на выпуск.
6.53. Должны быть разработаны письменные процедуры, которые необходимо соблюдать при расследовании критических отклонений или при несоответствии серий промежуточной продукции или АФС их спецификациям. Такое расследование должно распространяться и на другие серии, к которым могли бы иметь отношение данные несоответствия или отклонения.
6.6. Записи лабораторного контроля
6.60. Записи лабораторного контроля должны включать в себя полную информацию, полученную в ходе всех испытаний, проведенных для подтверждения соответствия установленным спецификациям и стандартам, включая исследования и количественные определения, в том числе:
описание образцов, полученных для проведения испытания, включая название исходного сырья или источника, номер серии или другой характерный код, дата отбора пробы, количество образца, представленного для проведения испытаний (при необходимости), и дата его получения;
описание каждого используемого метода испытаний или ссылка на такой метод;
указание массы образца или других единиц измерения для образца, используемого для каждого испытания, в соответствии с описанным методом, данные о приготовлении и испытании стандартных образцов, реактивов и стандартных растворов или соответствующие перекрестные ссылки;
записи всех исходных данных, получаемых в ходе каждого испытания, в дополнение к графикам, таблицам и спектрам, полученным с помощью лабораторных приборов, надлежащим образом идентифицированные для конкретного вещества и серии, подвергаемых испытанию;
записи всех расчетов, выполненных в связи с проведением испытания, включая, например, единицы измерения, коэффициенты пересчета и факторы эквивалентности;
результаты испытаний и их соответствие установленным критериям приемлемости;
подпись лица, проводившего каждое испытание, и дата (даты) его проведения;
дата и подпись второго лица, свидетельствующая, что оригиналы записей были проверены в отношении точности, полноты и соответствия установленным стандартам.
6.61. Следует также вести полные записи с указанием: любых изменений установленных аналитических методик; периодической калибровки лабораторного оборудования, аппаратов, средств измерений и регистрирующих устройств; всех исследований АФС на стабильность;
расследования отклонений результатов испытаний от спецификаций.
6.7. Обзор записей по производству и контролю качества серии
(досье на серию)
6.70. Для определения соответствия промежуточной продукции или АФС установленным спецификациям перед выдачей разрешения на выпуск серии или перед ее реализацией следует разработать письменные процедуры, которым необходимо следовать при обзоре и утверждении записей по производству и лабораторному контролю серий, включая упаковку и маркировку.
6.71. Записи по производству и лабораторному контролю критических стадий процесса подлежат проверке и подтверждению отделом (отделами) качества перед выдачей разрешения на выпуск или перед реализацией каждой серии АФС. Записи по производству и лабораторному контролю для некритических стадий процесса могут быть проверены квалифицированным персоналом производственного отдела или других подразделений в соответствии с процедурами, утвержденными отделом (отделами) качества.
6.72. Все отклонения, отчеты о расследованиях и отклонениях результатов от спецификаций следует оценивать в процессе обзора записей по производству и контролю серии (досье на серию) перед выдачей разрешения на выпуск этой серии.
6.73. Отдел (отделы) качества может передавать производственному отделу обязанности и полномочия в отношении выдачи разрешения на использование промежуточной продукции, за исключением тех случаев, когда продукция предназначена для поставки за пределы сферы контроля производителя.
7. Работа с материалами
7.1. Общий контроль
7.10. Должны быть приняты в форме письменного документа процедуры, описывающие приемку, идентификацию, помещение в карантин, хранение, обращение, отбор проб, проведение испытаний, а также одобрение или отклонение материалов.
7.11. Производители промежуточной продукции и (или) АФС должны иметь систему оценки поставщиков материалов, критических для качества.
7.12. Поставка материалов должна осуществляться поставщиками, утвержденными отделом (отделами) качества, в соответствии с согласованными спецификациями.
7.13. Если поставщик критических для качества материалов не является его производителем, то производитель промежуточной продукции и (или) АФС должен знать название и адрес производителя этих материалов.
7.14. Замену поставщика критических для качества материалов следует проводить в соответствии с разделом 13 настоящей части.
7.2. Приемка и карантин
7.20. При получении и перед приемкой каждый контейнер с материалами или группу контейнеров обследуют визуально на правильность маркировки (включая соответствие названия, используемого поставщиком с названием, используемым заказчиком, если они отличаются) и наличие повреждений контейнера, пломб, свидетельств постороннего вмешательства или контаминации. Материалы следует содержать в карантине до отбора проб, проверки или проведения испытаний и получения разрешения на их использование.
7.21. До смешивания поступивших материалов с имеющимися запасами (например, растворителями или запасами в накопительных бункерах) их следует обозначить как соответствующие требованиям к этим материалам, прошедшие необходимые испытания (если применимо) и разрешенные для использования. Необходимо разработать процедуры по предотвращению ошибочной выгрузки поступивших материалов в имеющийся запас.
7.22. Следует исключить возможность перекрестной контаминации поставки ангро, если она осуществляется в емкостях, не предназначенных специально для нее. Для подтверждения этого могут использоваться одно или несколько следующих доказательств:
наличие документа, подтверждающего очистку;
наличие документа с результатами испытания на наличие следов примесей;
аудит поставщика.
7.23. Большие емкости для хранения и обслуживающие их трубопроводы, линии наполнения и разгрузки должны иметь соответствующую маркировку.
7.24. Каждый контейнер или группу контейнеров с материалами (серией материалов) следует идентифицировать с помощью характерного кода, номера серии или номера, присвоенного при приемке. Этот номер следует использовать при регистрации местонахождения каждой серии. Должна быть система идентификации статуса каждой серии.
7.3. Отбор проб и проведение испытаний материалов, поступивших
для производства
7.30. Для подтверждения подлинности (идентификации) каждой серии материалов (за исключением материалов, указанных в пункте 7.32 настоящей части) следует провести хотя бы одно испытание. Если производитель имеет систему оценки поставщиков, вместо проведения других испытаний можно использовать сертификат анализа продукции поставщика.
7.31. Процедура утверждения поставщика должна включать в себя оценку способности производителя постоянно поставлять материалы, соответствующие спецификациям (например, данные о качестве предыдущих поставок). Прежде чем сократить объем испытаний при входном контроле, следует провести полный анализ как минимум 3 серий. Тем не менее следует проводить полный анализ через определенные промежутки времени и сравнивать его результаты с данными сертификата анализа поставщика. Достоверность данных сертификатов анализа следует регулярно проверять.
7.32. Не требуется проведение контроля технологических добавок, опасного или высокотоксичного сырья, других специальных материалов или материалов, передаваемых в другое подразделение под контролем заказчика, при предоставлении производителем продукции сертификата анализа, подтверждающего соответствие этих материалов установленным требованиям. Идентификация этих материалов проводится путем визуальной проверки контейнеров, этикеток и регистрации номеров серий. Отсутствие контроля таких материалов на месте производства лекарственного средства должно быть обосновано и оформлено документально.
7.33. Пробы должны быть репрезентативны для серии материалов, из которой они отобраны. В методиках отбора проб должны быть установлены количество контейнеров, из которых необходимо отбирать пробы, часть контейнера, откуда берется проба, а также количество материалов, которое должно быть отобрано из каждого контейнера. Число контейнеров для отбора проб и размер пробы должны быть указаны в плане отбора проб, в котором учитывается критичность материалов, изменчивость свойств материалов, предшествующий опыт работы с поставщиком в отношении качества, а также необходимое для анализа количество материалов.
7.34. Отбор проб необходимо проводить в определенных местах и в соответствии с инструкциями, предназначенными для предотвращения контаминации отобранной пробы и других материалов.
7.35. Контейнеры, из которых отбирают пробы, следует открывать осторожно и после отбора пробы немедленно снова закрывать. Их следует маркировать для указания факта отбора пробы.
7.4. Хранение
7.40. Обращение с материалами и их хранение следует осуществлять таким образом, чтобы предотвратить их разложение, контаминацию и перекрестную контаминацию.
7.41. Материалы, хранящиеся в фибровых барабанах, мешках или ящиках, не следует размещать на полу и располагать таким образом, чтобы дать возможность при необходимости провести очистку и осмотр.
7.42. Материалы следует хранить в течение периода и при условиях, которые не оказывают неблагоприятного воздействия на их качество. Как правило, необходимо следить, чтобы материалы, поступившие на хранение первыми, были использованы в первую очередь.
7.43. Некоторые материалы в соответствующих контейнерах могут храниться вне помещений при условии, что идентифицирующие этикетки остаются разборчивыми, а контейнеры надлежащим образом очищаются перед вскрытием и использованием.
7.44. Отклоненные материалы должны быть промаркированы и помещены под контроль в карантин, чтобы предотвратить их несанкционированное использование в производстве.
7.5. Повторная оценка
7.50. По мере необходимости материалы следует подвергать повторной оценке для того, чтобы определить их пригодность для использования (например, после продолжительного хранения или воздействия тепла или влажности).
8. Технологический процесс и контроль в процессе производства
8.1. Технологические операции
8.10. Исходное сырье для производства промежуточной продукции и АФС следует взвешивать или отмеривать в соответствующих условиях, не оказывающих влияния на его пригодность для использования. Весы и устройства для измерения объема должны иметь точность, приемлемую для предполагаемого использования.
8.11. Если исходное сырье делят на несколько порций для последующего использования в технологических операциях, контейнер, в который поступает исходное сырье, должен быть пригодным для этого и его маркировка должна содержать:
название исходного сырья и (или) его код;
номер, присвоенный при приемке, или контрольный номер;
масса или объем исходного сырья в новом контейнере;
дата проведения повторной оценки или повторных испытаний (при необходимости).
8.12. Следует установить наблюдение за критическими операциями взвешивания, отмеривания или разделения и удостоверять выполнение этих операций либо осуществлять их эквивалентный контроль. Перед использованием исходного сырья производственный персонал должен удостовериться в том, что оно действительно является тем сырьем, которое указано в записях по производству серии для данной промежуточной продукции или АФС.
8.13. Следует установить наблюдение или подвергать эквивалентному контролю и другие критические технологические операции.
8.14. На определенных стадиях технологического процесса фактические выходы следует сопоставлять с ожидаемыми выходами. Ожидаемые выходы и соответствующие пределы следует определять на основании данных проведенных ранее лабораторных, опытных или промышленных испытаний. Причины отклонений от ожидаемого выхода, связанные с критическими стадиями процесса, должны быть расследованы, чтобы определить их влияние (или возможное влияние) на качество соответствующих серий.
8.15. Любое отклонение должно быть оформлено документально с указанием причин отклонения. Любое критическое отклонение должно быть расследовано.
8.16. Технологический статус основных единиц оборудования следует указывать либо на конкретных единицах оборудования, либо в соответствующей документации, либо фиксировать в системе компьютерного контроля, либо с помощью альтернативных методов.
8.17. Необходимо надлежащим образом контролировать материалы, предназначенные для повторной обработки или переработки, с целью предотвращения их использования без соответствующего разрешения.
8.2. Ограничение времени выполнения операций
8.20. Если в технологической инструкции (пункт 6.41 настоящей части) установлены ограничения времени, их следует соблюдать для обеспечения качества промежуточной продукции и АФС. Отклонения от этих ограничений следует оформлять документально и проводить оценку отклонений. Указанные выше ограничения могут не вводиться при проведении технологического процесса до достижения установленных значений параметров (например, достижение необходимого значения рН, гидрогенизация, сушка до предварительно установленной величины параметра), если завершение реакций или стадий процесса определяется посредством отбора проб и испытаний в процессе производства.
8.21. Промежуточную продукцию, предназначенную для дальнейшей обработки, следует хранить при определенных условиях, чтобы обеспечить ее пригодность для дальнейшего использования.
8.3. Отбор проб и контроль в процессе производства
8.30. Следует разработать письменные инструкции по мониторингу процесса и контроля выполнения тех стадий процесса, которые являются причиной непостоянства показателей качества промежуточной продукции и АФС. Порядок проведения контроля в процессе производства и соответствующие критерии приемлемости определяют на основании информации, полученной на стадии разработки, или на основании опыта, приобретаемого в процессе производства.
8.31. Критерии приемлемости, тип и объем испытаний могут зависеть от природы выпускаемых промежуточной продукции и АФС, реакции или стадии процесса и степени влияния технологического процесса на непостоянство качества продукции. На начальных стадиях процесса допустимо проведение менее жесткого контроля в процессе производства, тогда как на более поздних стадиях процесса (например, стадии выделения и очистки) следует проводить более жесткий контроль.
8.32. Критические этапы контроля в процессе производства (и мониторинг критических процессов), включая точки и методы контроля, должны быть изложены в письменном виде и утверждены отделом качества.
8.33. Контроль в процессе производства может осуществлять квалифицированный производственный персонал. Корректировать данный процесс производства можно без предварительной санкции отдела качества, если такая корректировка не выходит за рамки заранее установленных пределов, утвержденных отделом качества. Все испытания и их результаты следует оформлять документально как часть записей на серию (досье на серию).
8.34. Методы отбора проб для материалов, использующихся в процессе производства, промежуточной продукции и АФС, должны быть зафиксированы в письменных инструкциях. Планы отбора проб и методики должны базироваться на научно обоснованном порядке отбора проб.
8.35. Отбор проб в процессе производства следует осуществлять с использованием процедур, предусматривающих предотвращение контаминации отобранного материала и другой промежуточной продукции или АФС. Необходимо разработать процедуры для обеспечения сохранения целостности образцов после отбора.
8.36. При проведении испытаний в процессе производства, которые осуществляются с целью мониторинга и (или) корректировки процесса, как правило, не требуется расследовать случаи несоответствия спецификациям.
8.4. Смешивание серий промежуточной продукции или АФС
8.40. В настоящей части понятие «смешивание» означает процесс объединения веществ для получения однородной промежуточной продукции или АФС, на которую выдается одна спецификация.
Смешивание в процессе производства частей одной и той же серии (например, объединение нескольких загрузок центрифуги из одной серии, полученной при кристаллизации) или объединение частей различных серий для последующей обработки считается частью технологического процесса и не рассматривается как смешивание.
8.41. Недопустимо смешивание в процессе производства серий веществ или продукции, не отвечающих требованиям спецификаций, с другими сериями таких веществ или продукции с целью обеспечения соответствия спецификациям. Каждая серия, входящая в состав смеси, должна быть произведена по установленной технологии, испытана отдельно от других серий и должна соответствовать установленным спецификациям перед смешиванием.
8.42. Операции смешивания приемлемы в частности для:
смешивания небольших серий для увеличения размера серии;
смешивания остатков (то есть относительно небольших количеств выделенного вещества) серий одной и той же промежуточной продукции или одной и той же АФС для получения единой серии.
8.43. Процессы смешивания следует надлежащим образом контролировать и оформлять документально. Полученную в результате смешивания серию при необходимости следует подвергать испытаниям на соответствие установленным спецификациям.
8.44. Записи, относящиеся к серии (досье на серию) и отражающие смешивание, должны обеспечивать возможность обратного прослеживания отдельных серий, из которых сделана смесь.
8.45. В тех случаях, когда физические характеристики АФС являются критическими (например, АФС, предназначенные для получения твердых лекарственных форм или суспензий для приема внутрь), операции смешивания следует валидировать, чтобы продемонстрировать однородность объединенной серии. Валидация должна включать проведение испытаний продукции по критическим характеристикам, на которые может оказать влияние процесс смешивания (например, распределение частиц по размерам, насыпной плотности и плотности при уплотнении).
8.46. Если смешивание может оказать отрицательное воздействие на стабильность, следует провести испытания стабильности окончательных серий, полученных в результате смешивания.
8.47. Дату окончания срока годности или дату проведения повторных испытаний серии, полученной в результате смешивания, следует определять на основании даты производства самых старых остатков или самой старой серии в смеси.
8.5. Контроль контаминации
8.50. Остатки веществ можно вносить в последующие серии той же промежуточной продукции или АФС при условии соответствующего контроля. Это могут быть остатки, налипшие на стенку измельчителя, слой влажных кристаллов, оставшихся на стенках бака центрифуги после разгрузки, и остатки, образовавшиеся в результате неполной выгрузки жидкостей или кристаллов из рабочей емкости при переносе вещества на следующую стадию процесса. Такое внесение не должно повлечь за собой переноса продуктов разложения или микробной контаминации, которые могут отрицательно повлиять на установленный профиль примесей АФС.
8.51. Технологические операции следует проводить таким образом, чтобы предотвратить контаминацию промежуточной продукции или АФС другими веществами.
8.52. С целью предотвращения контаминации следует соблюдать особые меры предосторожности при работе с АФС после ее очистки.
9. Упаковка и идентифицирующая маркировка АФС и промежуточной
продукции
9.1. Общие требования
9.10. Должны быть приняты в форме письменного документа процедуры, в которых описаны приемка, идентификация, помещение в карантин, отбор проб, исследование и (или) испытание и выдача разрешения на использование материалов для упаковки и маркировки, а также обращение с ними.
9.11. Материалы для упаковки и маркировки должны соответствовать установленным спецификациям. Материалы для упаковки и маркировки, не соответствующие таким спецификациям, должны быть отклонены для предотвращения их использования при выполнении операций, для которых они непригодны.
9.12. Следует вести записи для каждой поставки этикеток и упаковочных материалов с указанием данных об их приемке, проверке или испытании, а также об их принятии или отклонении.
9.2. Упаковочные материалы
9.20. Контейнеры (упаковки) должны обеспечивать надлежащую защиту от порчи или контаминации промежуточной продукции или АФС во время транспортирования и хранения промежуточной продукции или АФС в рекомендуемых условиях.
9.21. Контейнеры (упаковки) должны быть чистыми и, если этого требует характер промежуточной продукции или АФС, подвергаться санитарной обработке для обеспечения их пригодности для использования по назначению. Такие контейнеры (упаковки) не должны обладать химической активностью, абсорбирующими свойствами или служить источником посторонних примесей, чтобы не вызывать изменений качества промежуточной продукции или АФС сверх пределов, установленных в спецификации.
9.22. Если контейнеры (упаковки) предназначены для повторного использования, их следует очищать в соответствии с письменными инструкциями, а все предыдущие этикетки должны быть удалены или стерты.
9.3. Выдача этикеток и контроль
9.30. Доступ в зоны хранения этикеток должен быть разрешен только лицам, имеющим соответствующие полномочия.
9.31. Следует применять процедуры сопоставления количества выданных этикеток, количеству использованных и возвращенных этикеток, чтобы можно было оценить расхождения между количеством маркированных упаковок и количеством выданных этикеток. Факты расхождения необходимо расследовать, а полученные результаты должны быть утверждены отделом (отделами) качества.
9.32. Все неиспользованные этикетки с номерами серий или другой нанесенной информацией, относящейся к этим сериям, следует уничтожать. Возвращенные этикетки следует содержать и хранить таким образом, чтобы предотвратить перепутывание и обеспечить надлежащую идентификацию.
9.33. Устаревшие этикетки и этикетки с просроченными датами следует уничтожать.
9.34. Оборудование, используемое для изготовления этикеток при операциях упаковки, следует контролировать в целях обеспечения соответствия всех оттисков печатному тексту, указанному в записях по производству серии.
9.35. Отпечатанные этикетки, выдаваемые для определенной серии, следует тщательно проверять на предмет их подлинности и соответствия спецификациям, приведенным в основных записях производства. Результаты такой проверки следует оформлять документально.
9.36. Образец отпечатанной этикетки, соответствующий использованным этикеткам, следует включать в записи по производству серии.
9.4. Операции по упаковке и маркировке
9.40. Должны быть в наличии документально оформленные инструкции, предназначенные для обеспечения правильного использования упаковочных материалов и этикеток.
9.41. В операциях по маркировке должно быть предусмотрено предотвращение перепутывания. Необходимо физическое или пространственное разделение работ, связанных с разной промежуточной продукцией или АФС.
9.42. Этикетки, используемые для маркировки наружной поверхности контейнеров с промежуточной продукцией или АФС, должны содержать название или идентификационный код, номер серии продукции и условия хранения, если такая информация является критической для обеспечения качества промежуточной продукции или АФС.
9.43. Если промежуточная продукция или АФС предназначены для транспортировки за пределы сферы контроля системы управления материалами производителя, то на этикетке следует указать также наименование производителя и адрес производителя, количество содержимого, особые условия транспортирования и специальные требования, установленные законодательством государств-членов или актами органов Союза. Для промежуточной продукции и АФС, которые имеют установленный срок годности, на этикетке и в сертификате анализа следует указывать дату истечения срока годности. Для промежуточной продукции и АФС, для которых установлена дата повторных испытаний, эту дату следует указать на этикетке и (или) в сертификате анализа.
9.44. Помещения и оборудование для упаковки и маркировки следует проверить непосредственно перед их использованием, чтобы убедиться, что удалены все материалы, которые не нужны для следующей операции упаковки. Такая проверка должна быть оформлена документально в записях по производству серии, регистрационных журналах для помещений и эксплуатации оборудования или отражена в другой системе документального оформления.
9.45. Упакованную и маркированную промежуточную продукцию или АФС следует проверить, чтобы убедиться, что первичная и вторичная упаковка для серии имеют правильную маркировку. Эта проверка должна быть частью операции по упаковке. Результаты такой проверки должны быть отражены в записях по производству серии или документах по ее контролю.
9.46. Упаковки с промежуточной продукцией или АФС, подлежащие транспортированию за пределы сферы контроля производителя, следует опломбировать таким образом, чтобы в случае нарушения пломбы или ее отсутствия получатель мог обратить внимание на возможность изменения содержимого.
10. Хранение и реализация
10.1. Хранение на складе
10.10. Для хранения всех материалов при соответствующих условиях (например, контролируемые температура и влажность при необходимости) следует подготовить помещения и технические средства. Следует вести записи параметров этих условий, если они являются критическими для сохранения свойств материалов.
10.11. Если не имеется другой системы для предотвращения непреднамеренного или несанкционированного использования находящихся в карантине, отклоненных, возвращенных или отозванных материалов, то следует выделить отдельные зоны для временного хранения указанных материалов до принятия решения об их использовании.
10.2. Реализация
10.20. Реализация АФС и промежуточных продуктов третьим сторонам допускается только после выдачи отделом (отделами) качества разрешений на их выпуски, подтверждения соответствия серии уполномоченным лицом. АФС и промежуточная продукция, находящиеся в карантине могут быть переданы в другое подразделение, входящее в сферу контроля производителя, если это разрешено отделом (отделами) качества и при наличии необходимого контроля и соответствующей документации.
10.21. Условия транспортирования АФС и промежуточной продукции не должны оказывать отрицательного воздействия на их качество.
10.22. Особые условия транспортирования или хранения АФС или промежуточной продукции должны быть указаны на этикетке.
10.23. Производитель должен убедиться в том, что подрядчик, ответственный за перевозку АФС или промежуточной продукции, осведомлен о соответствующих условиях транспортирования и хранения и соблюдает их.
10.24. Должна быть внедрена система мер, позволяющая быстро установить пути реализации каждой серии промежуточной продукции и (или) АФС и осуществить их отзыв.
11. Лабораторный контроль
11.1. Общий контроль
11.10. Независимый отдел (отделы) качества должен иметь в своем распоряжении соответствующие лабораторные помещения и оборудование.
11.11. Должны быть в наличии письменные инструкции, описывающие отбор проб, проведение испытаний, одобрение или отклонение материалов, а также документальное оформление и хранение лабораторных данных. Ведение лабораторных записей должно соответствовать требованиям пункта 6.6 настоящей части.
11.12. Все спецификации, планы отбора проб и методики испытаний должны быть научно обоснованы и гарантировать то, что исходное сырье, промежуточная продукция, АФС, этикетки и упаковочные материалы соответствуют установленным стандартам качества и (или) чистоты. Спецификации и методики испытаний должны соответствовать требованиям регистрационного досье. Могут быть также спецификации, дополняющие спецификации регистрационного досье. Спецификации, планы отбора проб и методики испытаний, включая вносимые в эти документы изменения, должны быть составлены соответствующим подразделением, а также проверены и утверждены отделом (отделами) качества.
11.13. Следует разработать надлежащие спецификации для АФС, соответствующие принятым стандартам и согласующиеся с процессом производства. Спецификации должны включать контроль примесей (например, органических и неорганических примесей, остаточных растворителей). Если имеется спецификация для АФС в отношении микробиологической чистоты, то для общего количества микроорганизмов и при указании недопустимых микроорганизмов следует установить пределы, требующие принятия мер, и соблюдать эти требования. Если имеется спецификация для АФС в отношении содержания эндотоксинов, то следует установить соответствующие пределы, требующие принятия мер, и соблюдать эти требования.
11.14. Все процедуры лабораторного контроля должны проводиться в соответствии с утвержденными инструкциями и оформляться в письменном виде во время их выполнения. Любые отклонения от вышеуказанных процедур следует оформлять документально и объяснять.
11.15. Любые полученные данные о несоответствии показателей качества спецификации следует расследовать и документально оформлять согласно установленной процедуре. В соответствии с этой процедурой следует проанализировать данные, дать оценку того, имеются ли существенные проблемы, определить необходимые корректирующие действия и сделать выводы. Любой повторный отбор проб и (или) проведение повторных испытаний после получения результатов несоответствия спецификации следует выполнять согласно письменной процедуре.
11.16. Реактивы и стандартные растворы следует готовить и маркировать в соответствии с письменными инструкциями. На посуде с аналитическими реактивами или стандартными растворами (при необходимости) следует указывать дату, до которой они могут использоваться («использовать до...»).
11.17. При производстве АФС следует иметь (при необходимости) первичные стандартные образцы. Источник каждого первичного стандартного образца должен быть указан в документации. Необходимо вести записи хранения каждого первичного стандартного образца и его использования в соответствии с рекомендациями поставщика. Первичные стандартные образцы, полученные из официально признанных источников и хранящиеся в соответствии с рекомендациями поставщика, как правило, используют без проведения их испытаний.
11.18. При отсутствии первичного стандартного образца у официально признанного источника следует разработать «внутренний» первичный стандартный образец. Для достоверного установления подлинности и чистоты такого первичного стандартного образца следует провести надлежащие испытания. Необходимо сохранять соответствующую документацию проведения этих испытаний.
11.19. Вторичные стандартные образцы следует готовить, идентифицировать, испытывать, утверждать и хранить надлежащим образом. Перед первым использованием следует определять пригодность (провести первичную квалификацию) каждой серии вторичного стандартного образца путем сравнения с первичным стандартным образцом. Каждую серию вторичного стандартного образца следует периодически подвергать повторной квалификации в соответствии с письменным протоколом.
11.2. Испытания промежуточных продуктов и АФС
11.20. Для каждой серии промежуточной продукции и АФС следует проводить необходимые лабораторные испытания с целью подтверждения соответствия спецификациям.
11.21. Как правило, для каждого АФС следует устанавливать профиль примесей, описывающий идентифицированные и неидентифицированные примеси, присутствующие в типичной серии, полученной в результате определенного контролируемого технологического процесса. Профиль примесей должен включать идентификацию или другую качественную аналитическую характеристику (например, время удерживания), пределы содержания каждой обнаруживаемой примеси и классификацию каждой идентифицированной примеси (например, органическая и неорганическая примеси, растворитель). Профиль примесей, как правило, зависит от особенностей технологического процесса и происхождения АФС. Как правило, нет необходимости определять профиль примесей для АФС растительного или животного происхождения. Вопросы, связанные с биотехнологией, регулируются в соответствии с законодательством государств-членов и актами органов Союза.
11.22. Профиль примесей через определенные промежутки времени следует сравнивать с профилем примесей, приведенным в регистрационных документах, или с ранее полученными данными, чтобы обнаружить изменения в АФС, являющиеся результатом изменений сырья, параметров работы оборудования или технологического процесса.
11.23. Следует проводить соответствующие микробиологические испытания для каждой серии промежуточной продукции и АФС, если их микробиологическая чистота нормируется.
11.3. Валидация аналитических методик
(см. раздел 12 настоящей части)
11.4. Сертификаты анализа
11.40. Для каждой серии промежуточной продукции или АФС должен выдаваться оригинал сертификата анализа при его запросе.
11.41. Сертификат анализа должен содержать информацию о наименовании промежуточной продукции или АФС, включая при необходимости сорт, номер серии и дату выпуска. Если для промежуточной продукции или АФС установлена дата истечения срока годности, эту дату следует указывать на этикетке и в сертификате анализа. Если для промежуточной продукции или АФС установлена дата повторного испытания, то эту дату следует указывать на этикетке и (или) в сертификате анализа.
11.42. В сертификате должен быть приведен перечень всех испытаний, проведенных в соответствии с фармакопейными требованиями или требованиями потребителя, включая допустимые пределы, а также полученные количественные значения (если результаты испытаний таковыми являются).
11.43. Сертификат должен содержать дату подписания, подпись уполномоченного сотрудника отдела (отделов) качества, наименование, адрес и номер телефона первоначального производителя. Если анализ был проведен предприятием по переупаковке (повторной обработке), в сертификате анализа следует указать наименование, адрес, номер телефона предприятия по переупаковке (повторной обработке) и дать ссылку на наименование первоначального производителя.
11.44. Если новые сертификаты выдаются предприятием по переупаковке (повторной обработке), поставщиками или уполномоченными ими лицами, действующими от своего имени или от имени этого предприятия, то в них следует указывать наименование, адрес и номер телефона лаборатории, проводившей анализы.
Такие сертификаты должны также содержать ссылку на наименование и адрес первоначального производителя и первоначальный сертификат серии, копия которого прилагается к новому сертификату.
11.5. Контроль стабильности АФС
11.50. Необходимо принять в форме письменного документа программу продолжающихся испытаний, предназначенную для контроля стабильности характеристик АФС, полученные результаты испытаний необходимо использовать для подтверждения надлежащих условий хранения и дат проведения повторных испытаний или истечения срока годности АФС.
11.51. Методики испытаний, используемые при исследовании стабильности, должны пройти валидацию и обеспечивать получение необходимых данных о стабильности.
11.52. Образцы для испытания на стабильность следует хранить в контейнерах (упаковках), аналогичных коммерческим контейнерам (упаковкам). Например, если АФС реализуют в мешках, упакованных в фибровые барабаны, то образцы для испытаний на стабильность могут быть упакованы в мешки из того же материала, помещенные в барабаны меньшего размера, изготовленные из материала, аналогичного или идентичного материалу фибровых барабанов, в которых АФС поступает в продажу.
11.53. Как правило, в программу мониторинга стабильности для подтверждения дат проведения повторных испытаний или истечения срока годности должны быть включены первые 3 реализуемые производственные серии. Однако если данные предварительного исследования свидетельствуют, что АФС может сохранять стабильность по крайней мере в течение 2 лет, допускается использовать менее 3 серий.
11.54. В программу продолжающихся испытаний стабильности необходимо включать как минимум одну произведенную серию АФС в год (за исключением тех случаев, когда производственные серии в данном году не выпускались) и по крайней мере ежегодно проводить ее испытания для подтверждения стабильности.
11.55. Для АФС с короткими сроками хранения испытания необходимо проводить чаще. Например, для тех биотехнологических, биологических и других АФС, сроки хранения которых составляют 1 год или меньше, следует отбирать образцы для испытаний на стабильность и проводить испытания ежемесячно в течение первых 3 месяцев, а затем каждые 3 месяца. Если имеются данные, подтверждающие сохранение стабильности АФС, допускается исключить некоторые интервалы испытаний (например, 9-месячные испытания).
11.56. Необходимо, чтобы условия хранения при испытаниях на стабильность соответствовали требованиям законодательства государств-членов и актам органов Союза в отношении испытаний стабильности.
11.6. Даты истечения срока годности и проведения повторных испытаний
11.60. Если промежуточная продукция предназначена для передачи за пределы сферы контроля системы управления материалами производителя и для нее определена дата истечения срока годности или проведения повторных испытаний, то должна быть получена информация, подтверждающая стабильность промежуточной продукции (например, опубликованные данные, результаты испытаний).
11.61. Даты истечения срока годности или проведения повторных испытаний АФС должны основываться на результатах оценки данных, полученных при изучении стабильности. Общепринятой практикой является указание для АФС даты проведения повторных испытаний, а не даты истечения срока годности.
11.62. Предварительные даты истечения срока годности или проведения повторных испытаний АФС могут основываться на результатах, полученных для опытных серий, в следующих случаях:
если для опытных серий используются способ производства и процедуры, моделирующие окончательный процесс промышленного производства;
если качество АФС соответствует качеству вещества, которое будет выпускаться в промышленном масштабе.
11.63. Для проведения повторных испытаний следует отбирать репрезентативные образцы.
11.7. Архивные образцы
11.70. Архивные образцы упаковывают и хранят с целью возможной последующей оценки качества серий АФС, но не для проведения испытаний на стабильность.
11.71. Архивные образцы каждой серии АФС, надлежащим образом маркированные, следует хранить в течение 1 года после даты истечения срока годности серии, которая определяется производителем, или в течение 3 лет после реализации серии в зависимости от того, какой срок является более длительным. Архивные образцы АФС с установленной датой повторных испытаний следует хранить в течение 3 лет после того, как серия была полностью реализована производителем.
11.72. При хранении архивного образца следует использовать такую же систему контейнер (упаковка), в которой хранится АФС, или такую, которая эквивалентна системе упаковки, предназначенной для продажи, или обеспечивает лучшую защиту. Следует хранить архивные образцы в количестве, достаточном для проведения как минимум 2 полных анализов в соответствии с требованиями фармакопейной статьи или при отсутствии фармакопейной статьи - 2 полных анализов в соответствии со спецификацией (нормативным документом по качеству).
12. Валидация
12.1. Политика валидации
12.10. Производителем должна быть документально оформлена концепция в отношении поставленных целей и подхода к валидации, включая валидацию технологических процессов, процедур очистки, аналитических методик, процедур контроля в процессе производства, компьютеризированных систем, и в отношении лиц, ответственных за разработку, проверку, утверждение и документальное оформление каждого этапа валидации.
12.11. Критические параметры (показатели) качества, как правило, следует определять на стадии разработки или на основании данных предварительного опыта работы. Следует также определить диапазоны значений этих критических параметров (показателей) качества, необходимые для обеспечения воспроизводимости процесса. При этом необходимо:
определить критические показатели качества АФС как продукции;
указать параметры процесса, которые могут влиять на критические показатели качества АФС;
установить диапазон значений каждого критического параметра процесса, который предполагается использовать при серийном производстве и контроле процесса.
12.12. Операции, которые считаются критическими для качества и чистоты АФС, подлежат валидации.
12.2. Документация по валидации
12.20. Для каждого процесса, подлежащего валидации, следует разработать протокол валидации. Этот протокол должен быть проверен и утвержден отделом (отделами) качества и другими соответствующими службами.
12.21. В протоколе валидации должны быть определены критические стадии процесса и критерии приемлемости, а также вид проводимой валидации (например, ретроспективная, перспективная, сопутствующая) и количество производственных циклов.
12.22. Отчет о валидации должен содержать перекрестные ссылки на протокол валидации, обобщать полученные результаты и объяснять с соответствующими выводами любые обнаруженные отклонения, включая рекомендуемые изменения для исправления недостатков.
12.23. Любые отклонения от протокола валидации должны быть оформлены документально с соответствующим обоснованием.
12.3. Квалификация
12.30. До начала работ по валидации процесса следует завершить надлежащую квалификацию критического оборудования и вспомогательных систем. Квалификация обычно проводится по следующим этапам (по каждому этапу отдельно или по всем этапам в совокупности):
квалификация проекта (design qualification, DQ) - документально оформленное подтверждение того, что предложенный проект производственных помещений, оборудования или систем является пригодным для применения по назначению;
квалификация монтажа (installation qualification, IQ) - документально оформленное подтверждение того, что монтаж помещений, систем и оборудования (установленных или модифицированных) выполнен в соответствии с утвержденным проектом, рекомендациями производителя и (или) требованиями пользователя;
квалификация функционирования (operational qualification, ОQ) - документально оформленное подтверждение того, что помещения, системы и оборудование (установленные или модифицированные) функционируют в соответствии с предъявляемыми требованиями во всех предусмотренных режимах работы;
квалификация эксплуатации (performance qualification, PQ) - документально оформленное подтверждение того, что помещения, системы и оборудование при совместном использовании работают эффективно и воспроизводятся показатели, соответствующие утвержденным требованиям и характеристикам процесса.
12.4. Подходы к валидации процесса
12.40. Валидация процесса - это документально оформленное доказательство того, что процесс, протекающий в пределах установленных параметров, обеспечивает эффективное и с воспроизводимыми результатами производство промежуточной продукции или АФС, соответствующих предварительно заданным спецификациям и показателям качества.
12.41. Существует 3 подхода к валидации. Перспективная валидация является предпочтительным подходом, но имеются исключения, позволяющие использовать другие подходы. Эти подходы и их применимость описаны ниже.
12.42. Перспективную валидацию обычно следует выполнять для всех процессов, связанных с производством АФС, как указано в пункте 12.12 настоящей части. Перспективная валидация, проводимая для процесса, связанного с производством АФС, должна быть завершена до начала реализации готового лекарственного препарата, произведенного из этой АФС.
12.43. Сопутствующая валидация может быть проведена при отсутствии данных для повторяющихся технологических циклов, если выпущено ограниченное число серий АФС, если серии АФС выпускались редко или были произведены посредством валидированного процесса, который был модифицирован. До завершения сопутствующей валидации серии АФС могут быть выпущены и использованы для производства лекарственного препарата, предназначенного для реализации, при условии полного контроля и мониторинга серий АФС.
12.44. Исключением, позволяющим провести ретроспективную валидацию, являются достаточно организованные процессы, которые обеспечивают производство АФС без существенных изменений его качества вследствие изменений сырья, оборудования, систем, помещений и процесса производства. Такой подход к валидации может быть использован, если:
определены критические показатели качества и критические параметры процесса;
установлены надлежащие критерии приемлемости и контроля в процессе производства;
отсутствовали существенные сбои в ходе процесса или дефекты продукции по причинам, не связанным с ошибками оператора или отказами оборудования;
были установлены профили примесей для данной АФС.
12.45. Серии, отобранные для ретроспективной валидации, должны представлять собой репрезентативную выборку из всех серий, произведенных за проверяемый период, в том числе любых серий, не соответствующих спецификациям. При этом количество таких серий должно быть достаточным для доказательства постоянства процесса. В целях получения данных для ретроспективной валидации процесса может быть проведено испытание архивных образцов.
12.5. Программа валидации процесса
12.50. Количество производственных циклов, необходимых для валидации, должно зависеть от сложности процесса или значимости изменения процесса, подлежащих рассмотрению. Для перспективной и сопутствующей валидации должны быть использованы данные, полученные для 3 последовательных производственных серий продукции надлежащего качества. Однако могут быть ситуации, когда для доказательства постоянства процесса необходимы дополнительные производственные циклы (например, процессы производства сложных АФС или длительные процессы производства АФС). Для оценки постоянства процесса при ретроспективной валидации, как правило, необходимо исследовать данные для 10 - 30 последовательных серий, но при соответствующем обосновании это число может быть уменьшено.
12.51. Во время проведения исследований по валидации процесса следует контролировать и проверять его критические параметры. Параметры процесса, не связанные с качеством, например, переменные, контролируемые в целях сокращения потребления энергии или использования оборудования, можно не включать в валидацию процесса.
12.52. Валидация процесса должна подтверждать, что профиль примесей для каждого АФС находится в заданных пределах. Профиль примесей должен быть аналогичен (либо быть лучше) ранее полученному профилю примесей, а также в соответствующих случаях профилю примесей, установленному при разработке процесса, или серий, использованных для основных клинических и токсикологических исследований.
12.6. Периодическая проверка валидированных систем
12.60. Системы и процессы следует подвергать периодической оценке для подтверждения того, что они функционируют правильным образом. Если в процесс или систему не были внесены существенные изменения и обзор качества подтвердил, что система или процесс постоянно обеспечивает производство материала, соответствующего спецификациям, повторная валидация, как правило, не проводится.
12.7. Валидация очистки
12.70. Процедуры очистки, как правило, должны пройти валидацию. Обычно валидацию очистки проводят в случаях, при которых контаминация или перенос веществ представляет наибольшую опасность для качества АФС. Например, на начальных стадиях технологического процесса может не потребоваться проведение валидации процедур очистки оборудования, если остаточные вещества будут удалены на последующих стадиях очистки.
12.71. Валидация процедур очистки должна отражать фактический характер использования оборудования. Если разные АФС или разную промежуточную продукцию производят на одном и том же оборудовании и это оборудование очищают одним и тем же способом, то для валидации очистки можно выбрать репрезентативную промежуточную продукцию или АФС. Такой выбор должен основываться на данных о растворимости и трудностях очистки, а также на расчете предельного содержания остатков с учетом их активности, токсичности и стабильности.
12.72. В протоколе валидации очистки должны быть описаны оборудование, подлежащее очистке, процедуры, материалы, приемлемые уровни очистки, контролируемые и регулируемые параметры и аналитические методики. В протоколе следует также указать виды отбираемых проб, способы их отбора и маркировки.
12.73. Для обнаружения как нерастворимых, так и растворимых остатков методы отбора проб должны включать соответствующие методы (например, забор мазков или смывов, прямую экстракцию и др.). Используемые методы отбора проб должны позволять количественно определять уровни остатков на поверхностях оборудования после очистки. Следует учитывать, что использование метода отбора проб посредством забора мазков неосуществимо, если контактирующие с продуктом поверхности являются труднодоступными вследствие конструктивных особенностей оборудования (например, внутренние поверхности шлангов, транспортных трубопроводов, емкости реакторов с узкими люками, а также небольшое по размеру сложное оборудование (например, микронизаторы и микрораспылители)) и (или) если существуют ограничения процесса (например, обработка токсичных веществ), в этих случаях необходимо предусмотреть иной метод отбора проб.
12.74. Следует использовать валидированные аналитические методики, обладающие достаточной чувствительностью для обнаружения остатков или контаминантов. Предел обнаружения каждой аналитической методики должен быть достаточным для того, чтобы установить достижение приемлемого уровня остатков или контаминантов. Для методики следует установить достигаемый уровень извлечения вещества. Пределы содержания остатков должны быть практичными, достижимыми, проверяемыми и должны основываться на содержании наиболее вредного остатка. Пределы можно устанавливать, основываясь на минимальном обладающем известной фармакологической, токсикологической или физиологической активностью количестве АФС или наиболее вредного компонента АФС.
12.75. Для процессов, в которых существует необходимость снижения общего количества микроорганизмов или эндотоксинов в АФС, или для других процессов, где может быть значимым такая контаминация (например, производство нестерильных АФС, используемых для производства стерильных лекарственных препаратов), исследование очистки (санитарной обработки) оборудования следует проводить в отношении контаминации микроорганизмами и эндотоксинами.
12.76. Процедуры очистки следует контролировать с определенной периодичностью после валидации, чтобы убедиться, что эти процедуры являются эффективными при их использовании во время текущего технологического процесса. Чистоту оборудования можно контролировать посредством проведения аналитических испытаний и визуального осмотра. Визуальный осмотр позволяет обнаружить значительные скопления контаминантов на небольших участках, если есть основания полагать, что они могут оказаться не обнаруженными иным способом при отборе проб и (или) анализе.
12.8. Валидация аналитических методик
12.80. Если используемые аналитические методики не включены в Фармакопею Союза, фармакопеи государств-членов или в базовую фармакопею, определенную в соответствии с Концепцией гармонизации фармакопей государств-членов, а также другие стандарты, применяемые в рамках Союза, то они должны пройти валидацию.
Пригодность всех используемых методик испытаний следует проверять в реальных условиях применения и оформлять документально.
12.81. Валидацию методик следует проводить с учетом обоснования характеристик, приведенных в соответствующих актах органов Союза по валидации аналитических методик. Объем проводимой аналитической валидации должен зависеть от цели анализа и стадии технологического процесса производства АФС.
12.82. До начала валидации аналитических методик следует провести соответствующую квалификацию аналитического оборудования.
12.83. Следует вести подробные записи всех изменений валидированной аналитической методики. Такие записи должны отражать причину изменения и соответствующие данные для подтверждения того, что изменение приводит к результатам, обеспечивающим такую же точность и надежность аналитической методики, как и результаты, полученные с помощью ранее принятой аналитической методики.
13. Контроль изменений
13.10. Следует разработать формализованную систему контроля изменений для оценки всех изменений, которые могут повлиять на производство и контроль промежуточной продукции и АФС.
13.11. Следует предусмотреть письменные процедуры для идентификации, документального оформления, соответствующей проверки и утверждения изменений в отношении исходного сырья, спецификаций, аналитических методик, помещений, вспомогательных систем, оборудования (включая компьютерное оборудование), стадий процесса, материалов для маркировки и упаковки, а также компьютерного программного обеспечения.
13.12. Любые предложения по изменениям, касающимся соблюдения требований настоящих Правил, должны быть составлены, проверены и утверждены соответствующими подразделениями производителя, а затем проверены и утверждены отделом (отделами) качества.
13.13. Следует оценить возможное влияние предложенного изменения на качество промежуточной продукции или АФС. Процедура классификации изменений используется при определении объема испытаний, валидации и документации, требуемых для обоснования изменений, вносимых в ранее валидированный процесс. Изменения могут быть классифицированы (например, как существенные или несущественные) в зависимости от их характера и объема, а также влияния, которое они могут оказать на процесс. С учетом обоснованного заключения следует определить, какие дополнительные испытания и исследования по валидации необходимы для обоснования таких изменений.
13.14. При внедрении утвержденных изменений следует принять меры по пересмотру всех документов, на содержание которых влияют эти изменения.
13.15. После внедрения изменения в производство следует провести оценку первых серий, произведенных или испытанных после внедрения этого изменения.
13.16. Следует оценить возможность воздействия критических изменений на стабильность и на установленные даты повторных испытаний или даты истечения срока годности. При необходимости образцы промежуточной продукции или АФС, которые были произведены посредством измененного процесса, могут быть введены в программу ускоренного изучения стабильности и (или) включены в программу мониторинга стабильности.
13.17. Следует проинформировать соответствующих производителей лекарственных препаратов (в том числе лекарственных препаратов в форме нерасфасованных продуктов) об изменениях в установленных технологических процессах и процедурах контроля процесса, которые могут повлиять на качество АФС.
14. Отклонение и повторное использование материалов
14.1. Отклонение
14.10. Промежуточную продукцию и АФС, которые не соответствуют установленным спецификациям, следует содержать в условиях карантина, промаркировав так, чтобы обеспечить их явное отличие от продукции и АФС, соответствующих установленным спецификациям. Такую промежуточную продукцию или АФС можно подвергнуть повторной обработке или переработке. Окончательное решение в отношении отклоненных материалов должно быть оформлено документально.
14.2. Повторная обработка
14.20. Повторное использование в процессе производства промежуточной продукции или АФС, включая продукцию, не соответствующую стандартам или спецификациям, и их повторная обработка путем повторения стадии кристаллизации или других соответствующих стадий обработки химическими или физическими способами (например, дистилляция, фильтрация, хроматографирование, измельчение), являющихся частью установленного производственного процесса, обычно считаются приемлемыми. Однако если повторная обработка используется для большинства серий, то ее следует включить в качестве части стандартного технологического процесса.
14.21. Продолжение осуществления стадии технологического процесса, для которой контроль в процессе производства показал, что она не завершена, считается частью обычного технологического процесса, а не повторной обработкой.
14.22. Введение непрореагировавшего вещества в процесс вновь и повторное проведение химической реакции считаются повторной обработкой, если это не является частью установленного процесса. Такой повторной обработке должна предшествовать тщательная оценка, гарантирующая, что это не окажет негативного влияния на качество промежуточной продукции или АФС вследствие возможного образования побочных продуктов и веществ, прореагировавших сверх установленной нормы.
14.3. Переработка
14.30. Перед принятием решения о переработке серий, не соответствующих установленным нормам или спецификациям (нормативным документам по качеству), следует провести расследование причин такого несоответствия.
14.31. Серии, подвергнутые переработке, должны быть объектом соответствующей оценки, испытаний, исследования стабильности, если для этого есть основания, и документального оформления, чтобы показать, что переработанная продукция по качеству эквивалентна продукции, произведенной посредством первоначально установленного производственного процесса. Наиболее целесообразным подходом к валидации процедур переработки является сопутствующая валидация. Это позволяет составить записи процедуры переработки, установить порядок ее проведения и определить ожидаемые результаты. Если переработке подлежит только одна серия, то может быть составлен письменный отчет, а серия разрешена к выпуску сразу после подтверждения ее приемлемости.
14.32. Должны быть приняты в форме письменного документа процедуры для сравнения профиля примесей каждой переработанной серии с профилями примесей серий, произведенных посредством установленного процесса. Если применяемые аналитические методики не позволяют адекватно охарактеризовать переработанную серию, то следует воспользоваться дополнительными методиками.
14.4. Регенерация материалов и растворителей
14.40. Регенерация реактивов, промежуточной продукции или АФС (например, из маточной жидкости или фильтратов) считается допустимой при условии наличия утвержденных процедур регенерации и соответствия регенерированных материалов спецификациям, подходящим для их предполагаемого использования.
14.41. Допускаются регенерация и повторное использование растворителей в тех же или других процессах при условии, что процедуры регенерации контролируются и проверяются для обеспечения соответствия растворителей соответствующим стандартам перед их повторным использованием или смешиванием с другими материалами, допустимыми к использованию.
14.42. Новые и регенерированные растворители и реактивы можно смешивать, если в ходе соответствующих испытаний показана их пригодность для всех технологических процессов, в которых они могут использоваться.
14.43. Использование регенерированных растворителей, маточных жидкостей и других регенерированных веществ должно быть оформлено документально.
14.5. Возврат
14.50. Возвращенную промежуточную продукцию или возвращенные АФС следует маркировать соответствующим образом и содержать в условиях карантина.
14.51. Если состояние контейнеров (упаковки) или условия, в которых хранились или транспортировались промежуточная продукция или АФС до или в процессе их возврата, вызывают сомнения относительно качества промежуточной продукции или АФС, то возвращенная промежуточная продукция или возвращенные АФС подлежат повторной обработке, переработке или уничтожению.
14.52. Следует вести записи возврата промежуточной продукции или АФС. Для каждого возврата в документации следует указать:
наименование и адрес грузополучателя;
наименование промежуточной продукции или АФС, номер серии и возвращенное количество промежуточной продукции или АФС;
причину возврата;
указание на использование или уничтожение возвращенной промежуточной продукции или АФС.
15. Претензии и отзывы
15.10. Все претензии к качеству, полученные в устной или письменной форме, должны быть оформлены документально и расследованы согласно письменной инструкции.
15.11. Записи рассмотрения претензий должны содержать:
наименование, адрес и номер телефона лица, предъявившего претензию;
фамилия, имя, отчество (при наличии) лица, предъявившего претензию, его должность (при наличии);
суть претензии (включая наименование и номер серии АФС);
дату поступления претензии;
первоначально принятые меры с указанием даты их осуществления и лица, принявшего меры;
любые последующие действия, связанные с предъявленной претензией;
ответ, направленный лицу, предъявившему претензию (включая дату направления ответа);
окончательное решение, принятое в отношении серии или партии промежуточной продукции или АФС в связи с предъявленной претензией.
15.12. Записи рассмотрения претензий следует сохранять с целью оценки тенденций, частоты поступления претензий и их значимости для принятия дополнительного и при необходимости немедленного корректирующего действия.
15.13. Необходимо принять в форме письменного документа процедуру для определения обстоятельств, при которых следует рассматривать вопрос об отзыве промежуточной продукции или АФС.
15.14. Процедура отзыва должна определять, кто должен принимать участие в оценке информации, как следует начинать процедуру отзыва, кого следует проинформировать об отзыве промежуточной продукции или АФС и как следует поступать с отозванной промежуточной продукцией или АФС.
15.15. В случае серьезной или потенциально угрожающей жизни ситуации следует проинформировать об этом уполномоченные органы государств-членов, местные и (или) международные уполномоченные органы, а также при необходимости дальнейших совместных действий обратиться к ним за консультацией.
16. Производство по контракту
(включая лабораторный контроль качества)
16.10. Работающие по контрактам производители, а также осуществляющие контроль качества лаборатории, должны соблюдать требования настоящих Правил. Следует уделить особое внимание предотвращению перекрестной контаминации и обеспечению прослеживаемости.
16.11. Производитель должен проводить оценку конкретных операций, выполняемых на участках выполняющих по его заказу работы по контракту других производителей, а также осуществляющих контроль качества лабораторий, на соответствие требованиям настоящих Правил.
16.12. У производителя должен храниться оригинал заключенного в письменной форме договора (контракта или официального соглашения) между заказчиком и исполнителем, в котором подробно определены обязанности каждой из сторон по соблюдению требований настоящих Правил, включая мероприятия в отношении качества.
16.13. Контрактом должно быть предусмотрено право заказчика проводить аудит производства исполнителя на соответствие требованиям настоящих Правил.
16.14. Если разрешено выполнение работ по субподрядам, то исполнитель не должен передавать третьей стороне какую-либо часть доверенных ему по контракту работ без предварительной оценки и заключения дополнительных соглашений с производителем, по заказу которого выполняются работы.
16.15. Производственные и лабораторные записи следует хранить на участке, на котором выполнялись работы. Производителем должна быть организована возможность доступа к указанным записям.
16.16. Не допускается внесение изменений в технологический процесс, методики испытаний, спецификации или другие требования контракта, а также замена или модернизация оборудования, относящиеся к предмету контракта, без уведомления заказчика и утверждения им изменений.
17. Поставщики или лица, действующие от их имени
(агенты, брокеры и трейдеры), дистрибьюторы, предприятия
по переупаковке или перемаркировке
17.1. Область применения
17.10. Настоящий раздел относится к лицам и организациям, не являющимся первоначальным производителем, которые могут приобретать, переупаковывать, перемаркировывать, поставлять (продавать) и хранить АФС или промежуточную продукцию.
17.11. Все поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке должны соблюдать требования настоящих Правил.
17.2. Прослеживаемость реализованных АФС
и промежуточной продукции
17.20. Поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке должны обеспечивать полную прослеживаемость поставляемых ими АФС и промежуточной продукции. Для этого должны быть оформлены и сохранены следующие документы (документально зафиксированные данные):
наименование первоначального производителя;
адрес первоначального производителя;
заказы на поставку;
накладные (транспортные документы);
документация о приемке;
наименование или обозначение АФС или промежуточной продукции;
номер серии, присвоенный производителем;
записи транспортирования и реализации;
оригиналы сертификатов анализа, включая сертификаты, полученные после переупаковки или перемаркировки, а также сертификаты, полученные от первоначального производителя;
дата проведения повторных испытаний или дату истечения срока годности.
17.3. Управление качеством
17.30. Поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке должны создать, задокументировать и внедрить эффективную систему управления качеством, как указано в разделе 2 настоящей части, а также вести необходимую документацию.
17.4. Переупаковка, перемаркировка и хранение АФС
и промежуточной продукции
17.40. Переупаковку, перемаркировку и хранение АФС и промежуточной продукции следует осуществлять в соответствии с требованиями настоящих Правил, чтобы избежать перепутывания или утраты идентичности либо чистоты АФС или промежуточной продукции.
17.41. Переупаковку следует осуществлять в соответствующих условиях производственной среды для предотвращения контаминации или перекрестной контаминации.
17.5. Стабильность
17.50. Если АФС или промежуточную продукцию переупаковывают в контейнеры (первичную упаковку), тип которых отличается от используемого производителем АФС или промежуточной продукции, то необходимо исследовать стабильность для обоснования установленной даты истечения срока годности или проведения повторных испытаний.
17.6. Передача информации
17.60. Поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке должны передавать всю информацию от производителя АФС или промежуточной продукции потребителю и от потребителя - производителю АФС или промежуточной продукции в том случае, если она касается качества АФС или промежуточной продукции или решений, принятых уполномоченными органами в отношении данной АФС или промежуточной продукции.
17.61. Поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке и перемаркировке, поставляющие АФС или промежуточную продукцию потребителю, должны указывать наименование первоначального производителя АФС или промежуточной продукции и номер (номера) поставляемой серии.
17.62. Поставщик обязан также по запросу уполномоченных органов государств-членов представить информацию о первоначальном производителе АФС или промежуточной продукции. Первоначальный производитель может дать ответ уполномоченному органу непосредственно или через уполномоченных им посредников при наличии у них соответствующих полномочий).
17.63. Необходимо соблюдать специальные требования в отношении сертификатов анализа, указанные в пункте 11.4настоящей части.
17.7. Работа с претензиями и отзывами
17.70. Поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке должны вести записи рассмотрения претензий и отзывов, как указано в разделе 15 настоящей части, в отношении всех претензий и отзывов, которые относятся к сфере их деятельности.
17.71. При необходимости поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке должны рассматривать претензию совместно с первоначальным производителем АФС или промежуточной продукции, чтобы определить, следует ли предпринимать дальнейшие действия совместно с другими потребителями, которые могли получить такие же АФС или промежуточную продукцию, либо с уполномоченным органом, либо со всеми заинтересованными сторонами. Расследование причины претензии или отзыва должно проводиться соответствующей стороной, к сфере деятельности которой относятся претензия или отзыв, и оформляться документально.
17.72. Если претензия касается первоначального производителя АФС или промежуточной продукции, то записи рассмотрения претензии, которые ведут поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке, должны содержать все данные, полученные от первоначального производителя АФС или промежуточной продукции (включая дату и представленную информацию).
17.8. Работа с возвратами
17.80. Работу с возвратами продукции проводится в соответствии с требованиями пункта 14.52 настоящей части. Поставщики или лица, действующие от их имени, дистрибьюторы, предприятия по переупаковке или перемаркировке должны вести документацию по возвращенным АФС и промежуточной продукции.
18. Специальное руководство по АФС, производимым путем культивирования клеток или ферментации
18.1. Общие требования
18.10. В настоящем разделе рассматриваются специальные требования к контролю АФС или промежуточной продукции, которые производят посредством культивирования клеток или ферментации с использованием природных или рекомбинантных организмов, недостаточно отраженные в других разделах настоящих Правил. Этот раздел настоящих Правил не следует рассматривать отдельно от других разделов настоящих Правил, поскольку, как правило, требования, приведенные в других разделах настоящих Правил, также применимы к такой продукции. Следует отметить, что для «классических» процессов получения низкомолекулярных веществ и для процессов, в которых рекомбинантные и нерекомбинантные организмы используются для производства белков и (или) полипептидов, применяют одни и те же принципы ферментации, хотя степень контроля при этом будет различной. В настоящем разделе указаны эти различия, если они существуют на практике. Как правило, степень контроля биотехнологических процессов, используемых для производства белков и полипептидов, выше, чем для классических процессов ферментации.
18.11. Термин «биотехнологический процесс» относится к использованию клеток или организмов, полученных или модифицированных посредством технологии рекомбинантной ДНК, гибридомной или другой технологии, с целью производства АФС. АФС, полученные с помощью биотехнологических процессов, обычно состоят из таких высокомолекулярных соединений, как белки и полипептиды, специальное руководство в отношении которых приведено в настоящем разделе. По технологии рекомбинантной ДНК также могут быть получены такие определенные АФС с низкой молекулярной массой, как антибиотики, аминокислоты, витамины и углеводы. Уровень контроля этих видов АФС аналогичен контролю, применяемому для классической ферментации.
18.12. Термин «классическая ферментация» относится к процессам получения АФС, в которых используются природные микроорганизмы и (или) микроорганизмы, модифицированные общепринятыми методами (например, посредством облучения или химического мутагенеза). АФС, полученные посредством классической ферментации, обычно являются такими продуктами с низкой молекулярной массой, как антибиотики, аминокислоты, витамины и углеводы.
18.13. Производство АФС или промежуточной продукции из клеточных культур или методом ферментации включает в себя такие процессы, как культивирование клеток или экстрагирование и очистка материала, полученного от живых организмов. Следует отметить, что эти процессы могут включать такую дополнительную стадию, являющуюся частью производственного процесса, как физико-химическая модификация. Используемое сырье (среды, буферные компоненты) может обеспечивать возможность роста контаминирующих микроорганизмов. В зависимости от источника, способа получения и предполагаемого применения АФС или промежуточной продукции может быть необходим контроль бионагрузки, контаминации вирусами и (или) эндотоксинами во время производства и мониторинга процесса на соответствующих стадиях.
18.14. Для обеспечения качества промежуточной продукции и (или) АФС на всех стадиях производства следует установить надлежащий контроль. Хотя настоящая часть применяется начиная со стадии культивирования клеток или ферментации, предшествующие стадии (например, создание банка клеток) следует осуществлять при надлежащем производственном контроле. Настоящая часть охватывает культивирование клеток или ферментацию начиная с того момента, когда из банка клеток извлекают флакон с культурой клеток для использования в производстве.
18.15. Для сведения к минимуму риска контаминации следует использовать надлежащее оборудование и проводить контроль производственной среды. Критерии приемлемости для качества производственной среды и частота контроля зависят от стадии и условий технологического процесса (открытые, закрытые или изолированные системы).
18.16. Внутрипроизводственный контроль необходимо, как правило, распространить на следующее:
поддержание рабочего банка клеток (при наличии);
правильный посев и рост культуры;
контроль критических рабочих параметров во время культивирования клеток или ферментации;
контроль процесса роста клеток, их жизнеспособности (для большинства процессов культивирования клеток) и продуктивности (при необходимости);
процедуры сбора и очистки, при которых происходит удаление клеток, клеточных остатков и компонентов сред с одновременной защитой промежуточной продукции или АФС от контаминации (особенно контаминации микробиологической природы) и ухудшения качества;
контроль бионагрузки и уровней эндотоксинов на соответствующих стадиях технологического процесса (при необходимости);
вопросы вирусной безопасности в соответствии с законодательством государств-членов.
18.17. При необходимости, то следует доказать, что компоненты сред, белки клеток-хозяев, другие связанные с процессом и сопутствующие продукции примеси и контаминанты удалены.
18.2. Поддержание банка клеток и ведение записей
18.20. Доступ к банкам клеток должен быть разрешен только лицам, имеющим на это полномочия.
18.21. Банки клеток должны храниться в условиях, специально предназначенных для обеспечения поддержания жизнеспособности клеток и предотвращения контаминации.
18.22. Следует вести и сохранять записи использования и условий хранения флаконов из банков клеток.
18.23. При необходимости банки клеток следует периодически проверять с целью определения их пригодности для использования.
18.24. Более подробная информация, касающаяся поддержания банков клеток установлена соответствующими положениями законодательства государств-членов.
18.3. Культивирование клеток или ферментация
18.30. Если клеточные субстраты, среды, буферы и газы необходимо добавлять в асептических условиях, то по возможности следует использовать закрытые или изолированные системы. Если посев в первоначальной емкости или последующие переносы либо добавления сред, буферов выполняются в открытых емкостях, то следует осуществлять контроль и соответствующие процедуры для сведения к минимуму риска контаминации.
18.31. Если микробная контаминация может повлиять на качество АФС, то операции с использованием открытых емкостей следует проводить в боксе, обеспечивающем биологическую безопасность, или в производственной среде, контролируемой таким же образом.
18.32. При работе с культурами клеток персонал должен быть одет в специальную одежду и соблюдать специальные меры предосторожности.
18.33. Для обеспечения постоянства установленного процесса следует контролировать критические рабочие параметры (например, температуру, рН, скорость перемешивания, добавление газов, давление). Также следует контролировать рост, жизнеспособность (для большинства процессов культивирования клеток) и при необходимости продуктивность клеток. Критические параметры будут отличаться в зависимости от процессов и для классической ферментации может не потребоваться контроль определенных параметров (например, жизнеспособности клеток).
18.34. Оборудование, используемое для культивирования клеток, после использования должно быть очищено и стерилизовано. При необходимости оборудование для проведения ферментации также должно быть очищено, дезинфицировано или стерилизовано.
18.35. Питательные среды перед их использованием следует стерилизовать, если это целесообразно для предотвращения неблагоприятного влияния на качество АФС.
18.36. Для выявления контаминации и определения действий, которые необходимо осуществить, должны быть в наличии соответствующие процедуры, к которым относятся процедуры определения влияния контаминации на продукцию и процедуры деконтаминации оборудования и возвращения его к состоянию, позволяющему использовать это оборудование для производства последующих серий. Посторонние организмы, обнаруженные в ходе процессов ферментации, следует при необходимости идентифицировать и оценить их влияние на качество продукции. Результаты таких оценок следует принимать во внимание при решении вопроса о возможности использования полученного материала.
18.37. Следует сохранять записи случаев выявления контаминации.
18.38. После очистки универсального (предназначенного для производства многих видов продукции) оборудования между циклами по производству разной продукции может потребоваться проведение дополнительных испытаний с целью сведения к минимуму риска перекрестной контаминации.
18.4. Сбор, выделение и очистка
18.40. Стадии сбора как для удаления клеток или клеточных компонентов, так и для сбора клеточных компонентов после разрушения следует осуществлять с помощью оборудования и в зонах, предназначенных для сведения к минимуму риска контаминации.
18.41. Процедуры сбора и очистки, позволяющие удалять или инактивировать микроорганизм-продуцент, клеточные остатки и компоненты сред (при сведении к минимуму разрушения, контаминации и снижения качества), должны обеспечивать получение промежуточной продукции или АФС постоянного качества.
18.42. Все оборудование после использования должно быть очищено и подвергнуто санитарной обработке в установленном порядке. Производство нескольких последовательных серий промежуточной продукции и АФС без очистки оборудования допускается только в том случае, если это не оказывает влияния на их качество.
18.43. При использовании открытых систем очистка должна проводиться в контролируемых условиях производственной среды, обеспечивающих сохранение качества продукции.
18.44. Если оборудование используется для производства различных видов продукции, могут применяться такие дополнительные виды контроля, как использование специальных хроматографических смол или проведение дополнительных испытаний.
18.5. Стадии удаления или инактивации вирусов
18.50. Для получения более конкретной информации следует воспользоваться соответствующими нормативными правовыми актами государств-членов.
18.51. Стадии удаления и инактивации вирусов являются критическими стадиями обработки для некоторых процессов, и их следует осуществлять в пределах параметров, прошедших валидацию.
18.52. Следует принимать надлежащие меры предосторожности для предотвращения потенциальной контаминации вирусами продукции, прошедшей стадии удаления или инактивации вирусов, от продукции, которая эти стадии не прошла. Поэтому обработку в открытых системах следует осуществлять в зонах, отделенных от других этапов технологического процесса и имеющих отдельные системы обработки воздуха.
18.53. Одно и то же оборудование, как правило, не используется на различных стадиях очистки. Однако если необходимо использовать одно и то же оборудование, его перед повторным использованием следует надлежащим образом очистить и подвергнуть санитарной обработке. Следует предпринимать надлежащие меры предосторожности для предотвращения возможного переноса вирусов с предыдущих стадий (например, через оборудование или производственную среду).
19. АФС, предназначенные для
клинических исследований (испытаний)
19.1. Общие требования
19.10. Не все виды контроля, описанные в настоящей части, применимы при производстве оригинальных АФС, предназначенных для проведения исследований во время их разработки. В настоящем разделе приведены специальные требования к этим АФС.
19.11. Контроль, осуществляемый при производстве АФС, предназначенных для клинических исследований, должен соответствовать фазе разработки лекарственного препарата, в состав которого входит АФС. Процесс и методики исследований должны быть гибкими для обеспечения возможности внесения изменений по мере накопления знаний о процессе и продвижения исследований лекарственного препарата от доклинических исследований к клиническим исследованиям. Когда разработка лекарственного препарата достигает стадии, на которой АФС получают для применения в лекарственном препарате, предназначенном для клинических исследований, производители должны гарантировать, что АФС произведены с помощью надлежащих технических средств с использованием соответствующих процедур производства и контроля, необходимых для обеспечения качества АФС.
19.2. Качество
19.20. При производстве АФС, предназначенных для клинических исследований, необходимо применять соответствующие требования настоящих Правил и надлежащую процедуру одобрения каждой серии.
19.21. Необходимо организовать независимый от производства отдел (отделы) качества для одобрения или отклонения каждой серии АФС, предназначенной для клинических исследований.
19.22. Некоторые функции по проведению испытаний, выполняемых обычно отделом (отделами) качества, можно осуществлять в других подразделениях.
19.23. Мероприятия в отношении качества должны включать систему испытаний исходного сырья, упаковочных материалов, промежуточной продукции и АФС.
19.24. Следует анализировать проблемы, связанные с производством и качеством.
19.25. Текст маркировки АФС, предназначенных для клинических исследований, следует надлежащим образом контролировать: в нем должно быть указано, что вещество предназначено для исследовательских целей.
19.3. Помещения и оборудование
19.30. Во время всех фаз клинического исследования, включая использование опытных участков или лабораторий для производства серий АФС, предназначенных для клинических исследований, должны быть предусмотрены процедуры, гарантирующие, что оборудование прокалибровано, очищено и соответствует своему назначению.
19.31. Порядок эксплуатации оборудования должен гарантировать проведение работ с исходным сырьем таким образом, чтобы свести к минимуму риск контаминации и перекрестной контаминации.
19.4. Контроль исходного сырья
19.40. Исходное сырье, используемое при производстве АФС, предназначенных для клинических исследований, следует оценивать посредством проведения испытаний или получать вместе с результатами анализа, проведенного поставщиком, и проводить испытание на подлинность. Если вещество считается опасным, то достаточно анализа, проведенного поставщиком.
19.41. В некоторых случаях пригодность исходного сырья можно определять перед использованием на основании его пригодности при проведении реакции небольшого масштаба (то есть испытания функциональной пригодности), что предпочтительнее, чем только одни аналитические испытания.
19.5. Производство
19.50. Процесс производства АФС, предназначенных для клинических исследований, следует фиксировать в лабораторных журналах, в записях на серию или с помощью других подходящих средств. Эти документы должны включать информацию об использовании производственного сырья, оборудовании, технологическом процессе, а также научные наблюдения.
19.51. Ожидаемые выходы продукции могут различаться значительно и быть менее определенными, чем ожидаемые выходы продукции в процессах, выполняемых в промышленном масштабе.
Расследование причин отклонений от объема ожидаемого выхода не требуется.
19.6. Валидация
19.60. Если произведена одна серия АФС или изменения процесса во время разработки АФС делают воспроизводство серий затруднительным или неточным, валидация процесса производства АФС, предназначенной для клинических исследований, обычно нецелесообразна. На этой стадии разработки качество АФС обеспечивается сочетанием контроля, калибровки и при необходимости квалификации оборудования.
19.61. Если серии производят для коммерческого использования, даже при условии, что такие серии производят в опытном или опытно-промышленном масштабе, валидацию процесса следует проводить в соответствии с разделом 12 настоящей части.
19.7. Изменения
19.70. Изменения в технологическом процессе, спецификациях или методиках испытаний следует вносить во время разработки по мере приобретения новых знаний и роста масштаба производства. Каждое изменение следует надлежащим образом регистрировать.
19.8. Лабораторный контроль
19.80. Несмотря на то что аналитические методики, используемые для оценки серии АФС, предназначенной для клинических исследований, могут еще не пройти валидацию, они должны быть научно обоснованы.
19.81. Должна быть организована система хранения архивных образцов всех серий. Эта система должна обеспечивать сохранность достаточного количества каждого архивного образца в течение определенного периода после одобрения, завершения или отзыва заявки на регистрацию.
19.82. Определение даты истечения срока годности и проведения повторных испытаний, как указано в подразделе 11.6 настоящей части, применимо по отношению к существующим АФС, предназначенным для клинических исследований. Для новых АФС, находящихся на ранних стадиях клинических исследований, требования, определенные в указанном подразделе, обычно не применяются.
19.9. Документация
19.90. Система документации должна гарантировать, что информация, полученная в ходе разработки и производства АФС, предназначенных для клинических исследований, будет оформлена должным образом и доступна для использования.
19.91. Разработка и применение аналитических методик, используемых для подтверждения выпуска серии АФС, предназначенной для клинических исследований, должны быть оформлены документально.
19.92. Должна быть разработана и внедрена система хранения записей по производству и контролю и соответствующей документации. Эта система должна обеспечивать хранение записей и документов в течение установленного периода времени после одобрения, завершения или отзыва заявки на регистрацию.
20. Термины и определения
Для целей настоящей части используются основные понятия, которые означают следующее:
«активная фармацевтическая субстанция» (АФС, active pharmaceutical ingredient, API) - вещество или смесь веществ, используемые в производстве лекарственного препарата, которые в процессе производства становятся его активным ингредиентом (действующим веществом). Такие вещества обладают фармакологическим или другим непосредственным действием, предназначены для лечения, диагностики или профилактики заболеваний, ухода, обработки и облегчения симптомов, оказывают влияние на структуры или физиологические функции организма;
«бионагрузка» (bio burden) - уровень и вид микроорганизмов (например, неприемлемые или допустимые микроорганизмы), которые могут присутствовать в исходном сырье, исходном сырье для производства активной фармацевтической субстанции, промежуточной продукции или в активной фармацевтической субстанции. Бионагрузку не следует считать контаминацией, если ее уровни не превышают установленные предельные значения или не обнаружены микроорганизмы, определяемые как недопустимые;
«валидация» (validation) - документально оформленные действия, дающие высокую степень уверенности в том, что конкретный процесс, методика или система будут постоянно приводить к результатам, соответствующим заранее установленным критериям приемлемости;
«вспомогательные материалы» (process aids) - материалы, за исключением растворителей, являющиеся вспомогательными при производстве промежуточной продукции или активной фармацевтической субстанции и сами по себе не участвующие в химической или биологической реакции (например, фильтрующие материалы, активированный уголь и т. п.);
«выход ожидаемый» (yield, expected) - количество материала или процент от теоретического выхода, ожидаемые на любой соответствующей стадии технологического процесса, основанные на данных, предварительно полученных при производстве этого материала в лабораторных, опытных или промышленных условиях;
«выход теоретический» (yield, theoretical) - количество материала, которое определено на основании количества используемого материала и могло бы быть произведено на любой соответствующей стадии технологического процесса при условии отсутствия каких-либо потерь или отклонений в условиях реального технологического процесса;
«дата истечения срока годности» (expiry date or expiration date) - дата, указанная на упаковке (этикетках) активной фармацевтической субстанции и обозначающая период времени, в течение которого при хранении в установленных условиях характеристики активной фармацевтической субстанции должны оставаться в пределах спецификаций и по истечении которого активную фармацевтическую субстанцию нельзя использовать;
«дата повторного испытания» (retest date) - дата проведения повторного контроля материала для подтверждения его пригодности для дальнейшего использования;
«исходное сырье» (raw material) - общее понятие, используемое для обозначения исходных материалов, реактивов и растворителей, предназначенных для производства промежуточной продукции или активной фармацевтической субстанции;
«исходное сырье для производства активной фармацевтической субстанции» (API starting material) - сырье, промежуточная продукция или другие активной фармацевтической субстанции, которые используются для производства активной фармацевтической субстанции и входят в структуру активной фармацевтической субстанции в качестве важного структурного фрагмента. Исходное сырье для производства активной фармацевтической субстанции может быть закуплено у одного или нескольких поставщиков, либо может производиться самостоятельно. Исходное сырье для производства активной фармацевтической субстанции, как правило, имеет установленные химические свойства и структуру;
«калибровка» (calibration) - демонстрация того, что конкретный прибор или устройство дает результаты в установленных пределах по сравнению с результатами, получаемыми при использовании стандартного образца, или с результатами сопоставимого со стандартом образца во всем соответствующем диапазоне измерений;
«карантин» (quarantine) - статус веществ и материалов, изолированных физически или другими эффективными способами, до принятия решения об их последующем одобрении или отклонении;
«квалификация» (qualification) - действия, удостоверяющие и подтверждающие документально тот факт, что оборудование или вспомогательные системы смонтированы должным образом, правильно функционируют и действительно приводят к ожидаемым результатам. Квалификация является частью валидации, но отдельные этапы квалификации сами по себе не являются элементами валидации процесса;
«компьютеризированная система» (computerized system) - процесс или операция, объединенные в одно целое с компьютерной системой;
«компьютерная система» (computer system) - группа компонентов аппаратного обеспечения и соответствующего программного обеспечения, спроектированная и смонтированная таким образом, чтобы выполнять определенную функцию или набор функций;
«контаминация» (contamination) - нежелательное внесение примесей химической или микробиологической природы или инородных веществ в исходное сырье, промежуточную продукцию или активную фармацевтическую субстанцию во время технологического процесса, отбора проб, упаковки или переупаковки, хранения или транспортирования;
«контроль в процессе производства» (внутрипроизводственный контроль, межоперационный контроль) (in-process control or process control)) - проверки, осуществляемые в ходе технологического процесса с целью надзора (мониторинга) и при необходимости регулирования процесса и (или) для подтверждения того, что промежуточная продукция или активная фармацевтическая субстанция соответствуют спецификациям;
«контроль качества» (quality control, QC) - проверка или испытание на соответствие спецификациям;
«критерии приемлемости» (допустимые нормы) (acceptance criteria) - числовые пределы, интервалы или другие подходящие критерии приемлемости результатов испытаний;
«критический» (critical) - термин, относящийся к производственной стадии, условию технологического процесса, требованию испытаний или любому другому существенному параметру или предмету, которые следует поддерживать в рамках предварительно установленных критериев для обеспечения соответствия активной фармацевтической субстанции своей спецификации;
«лекарственный препарат» (drug (medicinal) product) - лекарственное средство в виде лекарственной формы;
«материал» (material) - общее понятие, обозначающее сырье (исходное сырье, реактивы, растворители), вспомогательные материалы, промежуточную продукцию, активную фармацевтическую субстанцию и материалы для упаковки и маркировки;
«маточная жидкость» (mother liquor) - остаточная жидкость после процессов кристаллизации или выделения. Маточная жидкость может содержать непрореагировавшие вещества, промежуточную продукцию, некоторые количества активной фармацевтической субстанции и (или) примесей. Она может быть использована для дальнейшей обработки;
«номер серии, номер партии» (batch number or lot number) - уникальная комбинация цифр, букв и (или) символов, которые идентифицируют серию (партию) и на основании которых можно определить историю ее производства и реализации;
«обеспечение качества» (quality assurance, QA) - совокупность всех организационных мероприятий, направленных на обеспечение того, чтобы все активные фармацевтические субстанции имели качество, необходимое для их предполагаемого применения, а все системы качества поддерживались в рабочем состоянии;
«отдел (отделы) качества» (quality unit(s)) - подразделение, независимое от производства и выполняющее обязанности как по обеспечению качества, так и по контролю качества. Это могут быть либо отдельные службы обеспечения качества и контроля качества, либо лицо или группа лиц в зависимости от масштаба и структуры организации;
«отклонение» (deviation) - отступление от утвержденной инструкции или утвержденного документа;
«перекрестная контаминация» (cross-contamination) - загрязнение материала или продукции другим материалом или продукцией;
«переработка» (reworking) - проведение одной или нескольких стадий, отличающихся от установленного производственного процесса, с целью обработки такой промежуточной продукции или активной фармацевтической субстанции, которая не соответствует стандартам или спецификациям, для получения промежуточной продукции или активной фармацевтической субстанции приемлемого качества (например, перекристаллизация с помощью другого растворителя);
«повторная обработка» (reprocessing) - возвращение в процесс промежуточной продукции или активной фармацевтической субстанции, включая продукцию, не соответствующую стандартам или спецификациям, и повторное проведение стадии кристаллизации или других соответствующих химических или физических стадий обработки (например, дистилляции, фильтрации, хроматографирования, измельчения), являющихся частью утвержденного производственного процесса. Продолжение осуществления стадии технологического процесса после того, как контроль в процессе производства показал, что стадия не завершена, считается частью обычного процесса, а не повторной обработкой;
«подписано (подпись)» (signed (signature)) - подпись лица, которое выполняло определенное действие или осуществляло проверку. Подпись может быть в виде инициалов, полного рукописного варианта имени и фамилии, рукописной подписи, личной печати или аутентичной и защищенной электронной подписи;
«примесь» (impurity) - любой компонент, присутствующий в промежуточной продукции или активной фармацевтической субстанции, наличие которого нежелательно;
«производитель, работающий по контракту» (contract manufacturer) - производитель, выполняющий определенный вид производственной деятельности по поручению первоначального производителя;
«производство» (manufacture) - операции и виды контроля, связанные с приемкой материалов, технологическим процессом, упаковкой, переупаковкой, маркировкой, перемаркировкой, выпуском, хранением и отгрузкой активной фармацевтической субстанции;
«промежуточная продукция» (intermediate) - материал, который получают в ходе стадий технологического процесса производства активной фармацевтической субстанции и который претерпевает дальнейшие молекулярные превращения или подвергается очистке, прежде чем станет активной фармацевтической субстанции. Промежуточная продукция в ходе технологического процесса может подвергаться или не подвергаться выделению;
«протокол валидации» (validation protocol) - документально оформленный план, указывающий, как следует проводить валидацию, и определяющий критерии приемлемости. Например, в протоколе валидации производственного процесса должны быть указаны технологическое оборудование, критические параметры процесса и его рабочие режимы, характеристики продукции, отбор проб, данные испытаний, которые необходимо собрать, количество валидационных циклов и приемлемые результаты испытаний;
«профиль примесей» (impurity profile) - описание идентифицированных и неидентифицированных примесей, присутствующих в активной фармацевтической субстанции;
«процедура» (procedure) - документально оформленное описание операций, подлежащих выполнению, мер предосторожности и мероприятий, прямо или косвенно относящихся к производству промежуточной продукции или активной фармацевтической субстанции;
«растворитель» (solvent) - неорганическая или органическая жидкость, используемая в качестве среды для приготовления растворов или суспензий при производстве промежуточной продукции или активной фармацевтической субстанции;
«серия (партия)» (batch (lot)) - конкретное количество материала, полученного в результате технологического процесса или серии процессов таким образом, что можно рассчитывать на его однородность в установленных пределах. В случае непрерывного производства серия может соответствовать определенной части продукции. Размер серии в этом случае может определяться либо фиксированным количеством, либо количеством, произведенным за определенный промежуток времени;
«спецификация» (specification) - перечень испытаний, ссылок на аналитические методики и критериев приемлемости, представляющих собой числовые границы, интервалы или другие критерии для соответствующих испытаний. Спецификация устанавливает набор критериев, которым должен соответствовать материал, чтобы считаться приемлемым для его предполагаемого применения. Соответствие спецификации означает, что материал, прошедший испытания согласно перечисленным аналитическим методикам, соответствует приведенным критериям приемлемости;
«стандартный образец, вторичный» (reference standard, secondary) - субстанция установленных качества и чистоты, что доказано посредством сравнения с первичным стандартным образцом, используемая в качестве стандартного образца для текущих лабораторных анализов;
«стандартный образец, первичный» (reference standard, primary) - субстанция, являющаяся подлинным веществом, что было доказано с помощью расширенных аналитических испытаний, и обладающая высокой степенью чистоты. Этот стандарт может быть получен из официально признанного источника или посредством независимого синтеза, или получен из используемого в производстве вещества с высокой степенью чистоты, или приготовлен посредством последующей очистки вещества, используемого в производстве;
«технологический процесс» (production) - все операции по производству активной фармацевтической субстанции, включая приемку материалов, обработку и упаковку активной фармацевтической субстанции;
«упаковочный материал» (packaging material) - любой материал, предназначенный для защиты промежуточной продукции или активной фармацевтической субстанции при хранении и транспортировании.
Часть III
Документы, связанные с Правилами надлежащей
производственной практики
Глава I. Пояснения по составлению
досье производственной площадки
1. Введение
1.1. Досье производственной площадки - документ, который составляется производителем лекарственных средств. Он должен содержать специальную информацию о политике в области качества и деятельности производственной площадки, технологическом процессе и (или) контроле качества при проведении на данной площадке операций по производству лекарственных средств, а также о каких-либо тесно взаимосвязанных работах в примыкающих и соседних зданиях. Если на данной площадке осуществляется только часть операций по производству, то в досье производственной площадки должны быть описаны только эти операции, например, анализ, упаковка и т. д.
1.2. При подаче досье производственной площадки в уполномоченный орган в нем должна быть представлена четкая информация о деятельности производителя в соответствии с требованиями настоящих Правил, которая может быть полезной при общем надзоре, а также для эффективного планирования и проведения инспектирования на соответствие требованиям настоящих Правил.
1.3. Досье производственной площадки должно содержать достаточную информацию, однако, насколько это возможно, его объем не должен превышать 25 - 30 страниц, не включая приложения. Простые планы, рисунки и схемы считаются более предпочтительными, чем описательное изложение. Досье производственной площадки (в том числе приложения) должно быть удобочитаемым при печати на бумаге формата А4.
1.4. Досье производственной площадки должно составлять часть документации, которая относится к системе управления качеством, его следует поддерживать в актуальном состоянии с целью отражения текущей деятельности. В досье производственной площадки необходимо указывать номер версии и дату введения в действие, а также дату следующего пересмотра. Каждое приложение может иметь отдельную дату введения в действие, что позволит осуществлять его независимый пересмотр.
2. Цель
Цель настоящего документа - дать рекомендации производителям лекарственных препаратов по созданию досье производственной площадки, которое может быть полезным для уполномоченного органа при планировании и проведении инспектирования производственной площадки на соответствие требованиям настоящих Правил.
3. Область применения
Положения настоящей главы применимы при подготовке и формировании содержания досье производственной площадки. Производители должны принимать во внимание региональные (национальные) нормативные правовые требования для определения того, является ли подготовка досье производственной площадки обязательным требованием для производителей лекарственных средств.
Положения настоящей главы распространяются на все виды производственной деятельности, такие как собственно производство, упаковка и маркировка, проведение испытаний, переупаковка или перемаркировка всех видов лекарственных препаратов. Основные принципы настоящих Правил могут использоваться при подготовке досье производственной площадки или соответствующего документа производителями лекарственных препаратов из донорской крови или тканей, а также производителями АФС.
4. Содержание досье производственной площадки
Досье производственной площадки должно содержать следующую информацию:
1. Общая информация о производителе.
1.1. Контактная информация: наименование и адрес юридического лица;
наименование(я) и фактический(ие) адрес(а) производственной(ых) площадки(ок), зданий и производственных участков, расположенных на этой(их) площадке(ах);
номер телефона персонала, работающего круглосуточно, с которым связываются в случае дефекта и (или) отзыва продукции;
идентификационный номер производственной площадки, например, координаты GPS или другой системы определения географического месторасположения (при наличии).
1.2. Информация о требующей специального разрешения (лицензии) производственной деятельности:
копия действующего специального разрешения (лицензии) на производство лекарственных средств, выданного уполномоченным органом (в качестве приложения 1);
краткое описание деятельности по производству, импорту, экспорту, оптовой торговле и пр., лицензированной соответствующими уполномоченными органами, включая зарубежные уполномоченные органы, с указанием лицензированных лекарственных форм (видов деятельности), если это не охватывается разрешением (лицензией) на производство;
виды продукции, производимой на площадке в настоящее время (перечень приводится в качестве приложения 2), если это не указано в приложении 1;
перечень инспекций площадки на соответствие требованиям надлежащей производственной практики за последние 5 лет, с указанием дат и названий (государств) уполномоченных органов, проводивших инспектирование, а также копия действующего сертификата соответствия требованиям надлежащей производственной практики (при наличии) (в качестве приложения 3 ).
1.3. Какая-либо другая производственная деятельность, осуществляемая на предприятии:
описание производственной деятельности на предприятии (производственной площадке), не связанной с фармацевтической деятельностью, если таковая проводится.
2. Система управления качеством производителя.
2.1. Система управления качеством производителя:
краткое описание системы управления качеством предприятия и ссылки на применяемые стандарты;
ответственность по поддержанию системы качества, включая высшее руководство;
информация о деятельности, в отношении которой предприятие аккредитовано и сертифицировано, включая даты и содержание документов по аккредитации (сертификации), наименования органов по аккредитации (сертификации).
2.2. Процедуры выдачи разрешения на выпуск готовой продукции: детальное описание квалификационных требований (образование и опыт работы) к уполномоченному(ым) лицу(ам), удостоверяющему соответствие серии установленным требованиям для выдачи разрешения на выпуск;
общее описание процедуры подтверждения соответствия серии установленным требованиям и выдачи разрешения на выпуск;
роль уполномоченного лица в выдаче разрешения на выпуск готовой продукции (в том числе снятие с карантина), а также в подтверждении соответствия требованиям регистрационного досье;
соглашения между уполномоченными лицами, если взаимодействуют несколько уполномоченных лиц;
указание на то, что в стратегии контроля используются процессно-аналитическая технология (РАТ) и (или) выпуск в реальном времени, или выпуск по параметрам (если они используются).
2.3. Управление поставщиками и подрядчиками: краткое резюме, содержащее информацию о цепях поставок, а также о программах внешнего аудита;
краткое описание системы квалификации подрядчиков, производителей активных фармацевтических субстанций и других поставщиков критических для качества материалов;
информация о мероприятиях по обеспечению соответствия продукции требованиям руководств в отношении губчатой энцефалопатии;
информация о мерах, предпринимаемых при подозрении или выявлении контрафактной, фальсифицированной, в том числе нерасфасованной продукции (например, неупакованных таблеток), активных фармацевтических субстанций или вспомогательных веществ;
информация об использовании внешней научной, аналитической или другой технической помощи, касающейся производства и анализа;
перечень контрактных производителей и лабораторий, включая адреса и контактную информацию, а также схемы цепей поставок для контрактной деятельности по производству и контролю качества, например, стерилизация первичного упаковочного материала для процессов в асептических условиях, испытания исходного сырья и т. д. (в качестве приложения 4);
краткий обзор распределения ответственности между заказчиком и исполнителем в отношении требований регистрационного досье (если не указано в подпункте 2.2 настоящего пункта).
2.4. Управление рисками для качества:
краткое описание используемой производителем методологии управления рисками для качества;
сфера действия и направленность управления рисками для качества, включая краткое описание любой деятельности, осуществляемой на корпоративном уровне, а также локальной деятельности. Должно быть упомянуто любое применение системы управления рисками для качества для оценки непрерывности поставок.
2.5. Обзоры качества продукции:
краткое описание примененной методологии.
3. Персонал.
Схема организационной структуры с указанием должностей (позиций) в управлении качеством, производстве и контроле качества, включая высшее руководство и уполномоченное(ые) лицо(а), (в качеств приложения 5);
количество персонала, занятого в управлении качеством, производстве, контроле качества, хранении и реализации.
4. Помещения и оборудование.
4.1. Помещения:
краткое описание предприятия, размер производственной площадки и перечень зданий (сооружений). Если производство осуществляется для различных рынков (например, локального, государств-членов, Европейского союза, Соединенных Штатов Америки и др.) в различных зданиях (сооружениях) производственной площадки, следует привести перечень этих зданий (сооружений) с указанием рынков, для которых предназначена производимая продукция (если это не указано в пункте 1.1);
простой план или описание производственных площадок (участков) с указанием масштаба (архитектурные и инженерные чертежи не нужны);
планы и схемы производственных зон (в качестве приложения 6); с указанием класса чистоты помещений и перепадов давления между прилегающими зонами, а также технологических операций (например, смешивание, наполнение, хранение, упаковка и т. д.), проводимых в помещениях;
планы складских помещений и зон хранения с обозначением специальных зон для хранения и обработки особо токсичных, опасных и сенсибилизирующих веществ (при наличии);
краткое описание не отмеченных в планах специальных условий хранения (при необходимости).
4.1.1. Краткое описание систем нагрева, вентиляции и кондиционирования воздуха (Heating, Ventilation, & Air Conditioning - HVAC):
принципы определения подачи воздуха, температуры, влажности, перепада давления и кратности обмена воздуха, уровень рециркуляции воздуха (в процентах).
4.1.2. Краткое описание систем водоподготовки: указание качества производимой воды; схематические чертежи систем (в качестве Приложения 7).
4.1.3. Краткое описание других систем обеспечения (системы подачи пара, сжатого воздуха, азота и т. п.).
4.2. Оборудование.
4.2.1. Перечень основного технологического и контрольного лабораторного оборудования с обозначением критических единиц (в качестве Приложения 8).
4.2.2. Очистка и дезинфекция:
краткое описание методов очистки и дезинфекции контактирующих с продукцией поверхностей (например, ручная очистка, автоматическая система «очистка на месте» и т. п.).
4.2.3. Описание компьютеризированных систем, критических с точки зрения требований настоящих Правил:
следует привести описание компьютеризированных систем, критических с точки зрения требований настоящих Правил, за исключением оборудования со специальными программируемыми логическими контроллерами.
5. Документация.
Описание системы документации (например, электронная, ручная).
Если документы и записи хранят или архивируют за пределами производственной площадки (включая данные по мониторингу безопасности лекарственных препаратов (при наличии)):
перечень видов документов (записей);
название и адрес иной площадки, где хранят документацию;
приблизительное время, необходимое для получения документов из архива, находящегося за пределами площадки.
6. Технологический процесс.
6.1. Виды продукции.
(Возможна ссылка на приложение 1 или 2)
а) Виды производимой продукции, включая:
перечень лекарственных форм как лекарственных препаратов для медицинского применения, так и ветеринарных лекарственных препаратов, производимых на площадке;
перечень лекарственных форм лекарственных препаратов, производимых на площадке для любых клинических исследований (следует представить информацию о производственных зонах и персонале, если они отличны от тех, что используются при серийном производстве);
б) Токсичные или опасные вещества (например, вещества с высокой фармакологической активностью и (или) сенсибилизирующими свойствами);
в) Виды продукции, производимой в специально предназначенных помещениях или на основе принципа кампаний (циклов производства) (при наличии);
г) Информация об использовании процессно-аналитической технологии (Process Analytical Technology - PAT) (при наличии):
общее описание соответствующей технологии и связанных с ней компьютеризированных систем.
6.2. Валидация процессов.
краткое описание общей политики в отношении валидации процессов;
описание политики в отношении повторной обработки и переработки.
6.3. Управление материалами и складское хранение:
информация о мероприятиях по обращению с исходным сырьем, упаковочными материалами, нерасфасованной и готовой продукцией, включая отбор проб, карантин, выдачу разрешения на выпуск и хранение;
информация о мероприятиях по обращению с отклоненными материалами и продукцией.
7. Контроль качества.
Следует привести описание деятельности по контролю качества, осуществляемой на производственной площадке в отношении физических, химических, микробиологических и биологических испытаний.
8. Дистрибьюция, претензии, дефекты продукции и отзывы.
8.1. Дистрибьюция (в части ответственности производителя):
типы организаций (держатели лицензий на дистрибьюцию, держатели лицензий на производство и т. д.), которым поставляется продукция производственной площадки, и их размещение (государства- члены, Европейский Союз, Соединенные Штаты Америки и т. д.);
описание системы, применяемой для подтверждения того, что каждый потребитель (получатель) имеет юридическое право получать от производителя лекарственные средства;
краткое описание системы обеспечения соответствующих условий во время перевозок (например, мониторинг (контроль) температуры);
описание организации дистрибьюции и методов, которыми обеспечивается прослеживаемость продукции;
описание мер по предупреждению попадания продукции производителя в незаконную цепь поставки.
8.2. Претензии, дефекты продукции и отзывы:
краткое описание системы работы с претензиями, дефектами продукции и по отзывам.
9. Самоинспекция:
краткое описание системы самоинспекций с особым вниманием к критериям выбора планово инспектируемых зон, практические мероприятия и дальнейшие действия.
Перечень необходимых приложений к досье производственной
площадки
Приложение 1. Копия действующей лицензии на производство.
Приложение 2. Перечень производимых лекарственных форм, в том числе международные непатентованные наименование или общепринятые названия (если существуют) используемых АФС.
Приложение 3. Копия действующего сертификата соответствия требованиям настоящих Правил.
Приложение 4. Перечень контрактных производителей и лабораторий, включая адреса и контактную информацию, а также схемы цепей осуществления этой контрактной деятельности.
Приложение 5. Организационные схемы.
Приложение 6. Планы производственных зон с указанием потоков исходного сырья и персонала, общая схема производственных процессов для каждого вида продукции (лекарственных форм).
Приложение 7. Схематические чертежи систем водоподготовки.
Приложение 8. Перечень основного технологического и лабораторного оборудования.
Глава II. Управление рисками для качества
Предисловие
Управление рисками для качества может применяться не только в производстве, но и по отношению к фармацевтической разработке, а также при составлении части регистрационного досье, касающейся качества. Этот документ могут применять уполномоченные органы при фармацевтической оценке части регистрационного досье, связанной с качеством, а также при проведении инспектирования на соответствие требованиям настоящих Правил и при расследованиях, связанных с предполагаемым дефектом.
Для обеспечения взаимосвязи с другими разделами данного документа данная глава включена в настоящую часть Правил. Что согласуется с положениями главы I части настоящих правил, которые содержат принципы управления рисками для качества.
Настоящая глава носит рекомендательный характер, в нем приводятся примеры процессов и применения управления рисками для качества.
1. Введение
Принципы управления рисками эффективно применяются во многих областях экономической деятельности и управления, включая финансы, страхование, безопасность на производстве, здравоохранение, мониторинг безопасности лекарственных препаратов, а также применяются уполномоченными органами, осуществляющими надзорную деятельность в этих областях. Хотя на сегодняшний день известны примеры применения управления рисками для качества в фармацевтической промышленности, они ограничены и не используют все возможности управления рисками. Кроме того, в фармацевтической промышленности признана важность систем качества и становится очевидным, что управление рисками для качества является важным компонентом эффективной системы качества.
Общепризнано, что риск определяется как комбинация вероятности причинения вреда и тяжести такого вреда. Однако сложно достичь однозначного понимания процесса управления рисками всеми заинтересованными сторонами, поскольку каждая из сторон может понимать возможный вред по-разному, вероятность возникновения вреда и характеристики его тяжести для каждого участника будут разными. В случае фармацевтической продукции, хотя и существуют различные заинтересованные стороны (в том числе пациенты, медицинские работники, правительственные органы и промышленность), первостепенное значение применения управления рисками для качества имеет защита пациента.
При производстве и применении лекарственного препарата, включая его компоненты, в определенной степени обязательно присутствуют риски. Риски для качества являются только одной составляющей общего риска. Важно понимать, что качество продукции следует поддерживать в течение жизненного цикла продукции таким образом, чтобы характеристики, имеющие значение для качества лекарственного препарата, оставались такими же, как у лекарственных препаратов, использовавшихся при клинических исследованиях. Эффективный подход к управлению рисками для качества может в дальнейшем гарантировать пациенту высокое качество лекарственного препарата с помощью установления в ходе разработки и производства предупреждающих методов идентификации и контроля возможных проблем, связанных с качеством. Кроме того, применение управления рисками для качества может усовершенствовать процедуру принятия решений в случае возникновения проблем с качеством. Эффективное управление рисками для качества может способствовать принятию лучших и более обоснованных решений, которые позволяют предприятию гарантировать уполномоченным органам эффективность решения вопросов, связанных с потенциальными рисками, а также благоприятно влиять на масштаб и уровень непосредственного контроля.
Цель данной главы - предложить системный подход к управлению рисками для качества. Это основополагающий документ производителя, который независим от других нормативных правовых актов в отношении качества (хотя и может быть связан, с ними) и который дополняет правила, требования, стандарты и руководства относительно качества, имеющиеся в фармацевтической промышленности и надзорной деятельности. Документ содержит специальные указания в отношении принципов и некоторых инструментов управления рисками для качества, что способствует принятию более эффективных и последовательных решений касательно рисков со стороны сотрудников как уполномоченных органов, так и предприятий в отношении качества активных фармацевтических субстанций и лекарственных препаратов в течение жизненного цикла продукции. Документ не устанавливает какие-либо новые требования в дополнение к действующим установленным требованиям.
Не всегда целесообразным и необходимым является формальный процесс управления рисками (с применением признанных способов и (или) внутренних процедур, например, стандартных операционных процедур). Считается приемлемым применение неформальных процессов управления рисками (с использованием эмпирических методов и (или) внутренних процедур). Надлежащее применение управления рисками для качества может облегчить выполнение обязанностей производителей (однако не отменяет их) в отношении соблюдения установленных требований, а также не заменяет соответствующий обмен информацией между представителями предприятий-производителей и уполномоченными органами.
2. Общие положения
В настоящей главе представлены принципы и примеры инструментов управления рисками для качества, которые могут быть применены к различным аспектам фармацевтического качества. Эти аспекты включают разработку, производство, оптовую торговлю, а также инспектирование и процессы представления заявок (обзоров) на протяжении жизненного цикла активных фармацевтических субстанций, лекарственных препаратов, биологических и биотехнологических лекарственных препаратов (включая использование исходного сырья, растворителей, вспомогательных веществ, упаковочных и маркировочных материалов для лекарственных препаратов, биологических и биотехнологических лекарственных препаратов).
3. Принципы управления рисками для качества
Существуют два основополагающих принципа управления рисками для качества:
оценка рисков для качества должна базироваться на научных данных и быть непосредственно связанной с защитой пациента;
уровень усилий, формализации и документального оформления процесса управления рисками для качества должен соответствовать уровню рисков.
4. Общий процесс управления рисками для качества
Управление рисками для качества - систематический процесс для общей оценки, контроля, информирования и обзора рисков для качества лекарственного препарата на протяжении его жизненного цикла. Модель управления рисками для качества представлена на рисунке 1 настоящего документа.
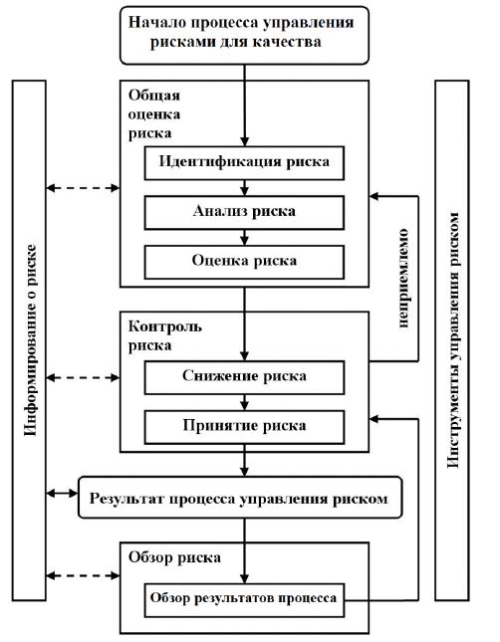
Рис. 1 Общая схема типового процесса управления рисками для качества.
Могут применяться и другие модели. Значение каждого компонента этой структуры может быть разным в разных случаях, но надежный процесс должен учитывать все компоненты, детализированные до такой степени, которая соответствует отдельному риску для качества.
На приведенной схеме не указаны точки принятия решений, поскольку решения могут быть приняты в любой точке процесса. Эти решения могут возвращать на предыдущий этап для поиска дальнейшей информации, чтобы откорректировать модели рисков для качества или даже прекратить процесс управления риском для качества из-за информации, являющейся основанием для такого решения. (Примечание: «неприемлемо» на рис. 1 настоящего раздела касается не только требований законодательства государств-членов и права Союза, но также необходимости пересмотреть процесс общей оценки рисков.)
4.1. Обязанности
Деятельность по управлению рисками для качества, как правило, осуществляется группами, включающими специалистов разных областей знаний. При формировании групп в них следует включать экспертов в соответствующих областях (например, отдела качества, развития бизнеса, инжиниринга, регуляторной деятельности, технологии, продаж и маркетинга, юридической службы, статистики и клиники), в дополнение к лицам, владеющим знаниями процесса управления рисками для качества.
Лица, ответственные за принятие решений, должны:
нести ответственность за координацию управления рисками для качества между разными видами деятельности и разными подразделениями в организации;
гарантировать, что процесс управления рисками для качества определен, находится в действии и проверяется и что имеются достаточные ресурсы.
4.2. Начало процесса управления рисками для качества
Управление рисками для качества должно включать систематические процессы, предназначенные для координации, облегчения и совершенствования принятия научно обоснованных решений в отношении рисков. Этапы, применяемые для планирования и начала выполнения процесса управления рисками для качества, могут включать:
определение проблемного и (или) представляющего риск вопроса, включая соответствующие предположения, устанавливающие возможность риска для качества;
сбор исходной информации и (или) данных о потенциальной опасности, вреде или влиянии на здоровье человека, имеющих отношение к общей оценке рисков;
определение руководителя и необходимых ресурсов; создание графика, связывающего уровень принятия решения с возможностью осуществления процесса управления рисками для качества.
4.3. Общая оценка рисков
Общая оценка рисков состоит из идентификации опасностей, а также анализа и оценки рисков, связанных с воздействием этих опасностей (как указано ниже). Общую оценку рисков для качества начинают с четкого описания проблемы или аспекта риска. Если рассматриваемый риск для качества четко определен, будет легче установить соответствующий инструмент управления риском (см. примеры в разделе 5 настоящей главы), а также виды информации относительно аспекта риска. Для четкого определения рисков в целях общей оценки рисков, как правило, применяются 3 основных вопроса:
что может происходить неверно;
какова вероятность (возможность) того, что это будет происходить неверно;
каковы последствия (их тяжесть).
Идентификация риска - систематическое использование информации для установления опасностей относительно аспекта риска или для описания проблемы. Информация должна включать предшествующие данные, теоретический анализ, мотивированные заключения, а также вопросы заинтересованных сторон.
Идентификация риска связана с ответом на вопрос: «что может происходить неверно?» и с установлением возможных последствий. Это дает основу для последующих этапов процесса управления рисками для качества.
Анализ риска - оценка риска, связанная с идентификацией опасностей. Это процесс установления качественной и количественной связи между вероятностью происшествия и тяжестью вреда. Для некоторых инструментов управления рисками возможность определить опасность (способность к выявлению) также является фактором оценки риска.
Оценка риска - сравнение идентифицированного и проанализированного риска с установленными критериями приемлемости риска. При оценке риска рассматривают обоснованность доказательств по всем 3 основным вопросам.
При общей оценке рисков важна обоснованность набора данных, поскольку это определяет качество результата. Выявление допущений и возможных причин неопределенности будет повышать правильность результата и (или) поможет определить ограничения. Неопределенность является следствием неполных знаний о процессе, а также его ожидаемой или неожидаемой вариабельности. Обычными причинами неопределенности является недостаток знаний в фармацевтической области и недостаточное понимание процесса, причины вреда (например, неправильные режимы процесса, причины вариабельности), а также недостаточная возможность выявления проблем.
Результатом общей оценки рисков является либо количественная оценка рисков, либо качественное описание диапазона рисков. Если риски выражены количественно, используют числовое выражение вероятности. Возможна также выражение риска с использованием качественных признаков, таких как «высокий», «средний» или «низкий», которые должны быть определены настолько подробно, насколько это возможно. Иногда используют «шкалу рисков» для дальнейшего определения признаков при ранжировании рисков. При выполнении количественной общей оценке рисков предусматривается вероятность специфического последствия, представленного как совокупность обстоятельств, способствующих возникновению риска. Таким образом, количественная оценка рисков применяется в отношении одного конкретного последствия, которое произошло в это время. Для некоторых инструментов управления рисками возможно также использование относительной меры риска в комбинации с множественными уровнями тяжести и вероятности для общей оценки относительного риска. На промежуточных этапах процесса установления шкалы можно применять количественную оценку рисков.
4.4. Контроль рисков
Контроль рисков предполагает принятие решения по снижению и (или) принятию рисков. Целью контроля рисков является снижение рисков до приемлемого уровня. Количество приложенных для контроля рисков усилий должно быть пропорционально важности рисков. Для понимания оптимального уровня рисков лица, ответственные за принятие решения, могут использовать разные процессы, в том числе анализ «Выгода-Затраты».
Контроль рисков должен сосредоточиться на следующих вопросах:
превышает ли риск приемлемый уровень; что может быть сделано для снижения или устранения риска; каков приемлемый баланс между выгодой, рисками и ресурсами; возникают ли новые риски в результате проведения контроля установленных рисков.
Снижение рисков сосредоточено на процессах уменьшения или устранения рисков для качества при превышении установленного (приемлемого) уровня риска (см. рис. 1). Снижение рисков может включать меры, предпринимаемые для уменьшения тяжести и вероятности вреда. Как часть стратегии контроля рисков могут использоваться процессы, улучшающие способность к выявлению опасности и рисков для качества. Внедрение мероприятий по снижению рисков может приводить к внесению новых рисков в систему или к возрастанию значимости других существующих рисков. Таким образом, после внедрения процесса снижения рисков может быть целесообразным пересмотреть общую оценку рисков для установления и оценки какого- либо возможного изменения рисков.
Принятие рисков - решение принять риски. Принятие рисков может быть формальным решением принять окончательные риски или пассивным решением, если окончательные риски не установлены. Относительно некоторых видов вреда даже наилучшие правила управления рисками для качества не в состоянии полностью устранить риски. При таких условиях может быть принято решение о том, что используется соответствующая стратегия управления рисками для качества и что риски для качества снижены до установленного (приемлемого) уровня. Такой установленный (приемлемый) уровень будет зависеть от многих параметров и должен быть определен в каждом отдельном случае.
4.5. Информирование о рисках
Информирование о рисках - передача информации относительно рисков и управления рисками лицам, ответственным за принятие решения, и другим заинтересованным лицам. Стороны могут быть проинформированы на любой стадии процесса управления рисками (см. рис. 1 настоящего документа, обозначено пунктирными стрелками). Следует надлежащим образом информировать о результатах процесса управления рисками для качества и оформлять их документально (см. рис. 1 настоящего документа, обозначено непрерывной стрелкой). Должен быть обмен информацией между всеми заинтересованными сторонами (например, между представителями уполномоченных органов и производителями, между представителями производителей и пациентом, между внутренним персоналом производителя, представителями производителей и уполномоченного органа и т. п.). Включенные сведения могут касаться существования, характера, формы, вероятности, тяжести, приемлемости, контроля, рассмотрения, способности к выявлению и других аспектов рисков для качества. Нет необходимости информировать о каждом случае принятия рисков. Информирование о решении в отношении управления рисками для качества между производителями и уполномоченными органами может эффективно осуществляться по имеющимся каналам, в соответствии с установленными нормативными правовыми актами и руководствами.
4.6. Обзор рисков
Управление рисками должно быть постоянно действующей частью процесса управления качеством. Следует ввести механизм обзора или мониторинга событий.
Результаты процесса управления рисками следует пересматривать с учетом новых знаний и опыта. Если процесс управления рисками для качества был начат, его следует продолжать, для того чтобы рассматривать события, которые могут повлиять на предыдущее решение в рамках процесса управления рисками для качества, независимо от того, являются ли эти события запланированными (например, обзор качества продукции, инспекция, аудит, контроль изменений) или незапланированными (например, основная причина при расследовании несоответствия, отзыв). Частота любого обзора должна основываться на уровне рисков. Обзор рисков может включать пересмотр решения о принятии рисков (пункт 4.4 данного раздела).
5. Методология управления рисками
Управление рисками для качества основывается на научном и практическом подходе к принятию решений. Оно предусматривает документально оформленные, понятные и воспроизводимые методы по осуществлению этапов процесса управления рисками для качества на основании имеющихся знаний относительно оценки вероятности, тяжести и иногда способности к выявлению рисков.
Традиционно оценку рисков для качества и управление ими осуществляли с помощью разных неформальных способов (например, эмпирических и (или) внутренних методик), которые базировались, например, на комбинации наблюдений, тенденций и другой информации. Эти подходы обеспечивают полезной информацией, что может оказать помощь в решении таких вопросов, как работа с претензиями, дефекты, отклонения, распределение ресурсов.
Кроме того, представители фармацевтической промышленности и уполномоченных органов могут оценивать риски и управлять ими с помощью признанных инструментов управления рисками и (или) внутренних процедур (например, стандартных операционных процедур). Перечень некоторых инструментов управления рисками (детальную информацию см. в Дополнении № 1 к настоящим Правилам и в разделе 8 настоящей части) включает в себя:
основные вспомогательные методы управления риском (блок- схемы, контрольные карты и тому подобное);
анализ режимов и последствий отказов (Failure Mode Effects Analysis - FMEA);
анализ режимов, последствий и критичности отказов (Failure Mode, Effects and Criticality Analysis - FMECA);
анализ древа ошибок (Fault Tree Analysis - FTA);
анализ опасностей и критические контрольные точки (Hazard Analysis and Critical Control Points - HACCP);
анализ опасности и работоспособности (Hazard Operability Analysis - HAZOP);
предварительный анализ опасности (Preliminary Hazard Analysis - PHA);
ранжирование и фильтрация рисков;
соответствующие статистические методы.
6. Внедрение управления рисками для качества в
промышленность и надзорную деятельность
Управление рисками для качества является процессом, способствующим принятию научно обоснованных и практических решений при его интеграции в системы качества (см. дополнение 2 к настоящим Правилам). Как указано выше, надлежащее применение управления рисками для качества не устраняет обязанности представителей промышленности соблюдать требования, установленные уполномоченными органами. Однако эффективное управление рисками для качества может способствовать принятию лучших и более обоснованных решений, что предоставит представителям уполномоченных органов большую гарантию способности предприятия управлять потенциальными рисками, а также может повлиять на масштаб и уровень непосредственного контроля со стороны уполномоченного органа. Кроме того, управление рисками для качества может способствовать лучшему использованию ресурсов всеми сторонами.
Обучение процессам управления рисками для качества как работников промышленности, так и представителей уполномоченных органов обеспечивает лучшее понимание процессов принятия решений и создает доверие к результатам управления рисками для качества.
Управление рисками для качества следует внедрять в существующую деятельность и должным образом оформлять документально. В дополнении № 2 к настоящим Правилам представлены примеры ситуаций, когда применение процесса управления рисками для качества может обеспечить информацией, которая может быть использована в различной деятельности в фармацевтической сфере. Эти примеры приведены с целью иллюстрации, не могут рассматриваться как окончательный и исчерпывающий перечень и не предназначены для установления каких-либо новых требований в дополнение к требованиям, установленным законодательством.
Примеры производственной деятельности и деятельности уполномоченных органов (см. Дополнение 2 к настоящим Правилам): управление качеством.
Примеры производственных операций и деятельности (см. Дополнение 2 к настоящим Правилам): разработка;
помещения, оборудование и системы обеспечения; управление исходным сырьем и материалами; производство;
лабораторный контроль и испытание стабильности; упаковка и маркировка;
Примеры деятельности уполномоченных органов (см. Дополнение 2 к настоящим Правилам):
инспектирование и оценка деятельности предприятия.
Поскольку решения уполномоченных органов принимаются отдельно в каждом государстве (регионе), общее понимание и применение принципов управления рисками для качества может способствовать взаимному доверию и принятию более согласованных решений представителями разных уполномоченных органов на основании одинаковой информации. Такое сотрудничество может быть важным при разработке политики и руководящих документов, которые вводят практики управления рисками для качества и способствуют их внедрению.
7. Термины и определения
Для целей настоящей части используются основные понятия, которые означают следующее:
«анализ рисков» (risk analysis) - оценка рисков в связи с установленной опасностью;
«вред» (harm) - ущерб, нанесенный здоровью человека, в том числе вред, являющийся следствием утраты качества продукции или ее пригодности;
«жизненный цикл продукции» (product lifecycle) - все фазы жизни продукции от начальной разработки, пребывания в обороте и до прекращения существования продукции;
«заинтересованная сторона» (stakeholder) - какое-либо лицо, группа лиц или организация, которые могут влиять на риски либо на которые могут влиять риски, либо которые считают себя находящимися под влиянием рисков. Лица, ответственные за принятие решений, также могут быть заинтересованной стороной. В настоящей части первостепенными заинтересованными сторонами являются пациент, медицинский работник, уполномоченный орган и предприятие- производитель;
«идентификация риска» (risk identification) - систематическое использование информации для выявления потенциальных источников вреда (опасности) со ссылкой на рассмотрение риска или описание проблемы;
«информирование о рисках» (risk communication) - передача информации о рисках и управлению рисками между лицом, ответственным за принятие решения, и другими заинтересованными сторонами;
«качество» (quality) - степень соответствия требованиям характеристик продукции, системы или процесса;
«контроль рисков» (risk control) - действия по выполнению решений по управлению рисками;
«лицо(а), ответственное(ые) за принятие решений» (decision maker(s)) - лицо(а), имеющее(ие) соответствующую компетенцию и полномочия для принятия надлежащих и своевременных решений по вопросам управления рисками для качества;
«обзор рисков» (risk review) - обзор или мониторинг итогов (результатов) процесса управления рисками с учетом (при необходимости) новых знаний и опыта о них;
«общая оценка рисков» (risk assessment) - систематический процесс формирования информации для обеспечения принятия решений в отношении рисков в рамках процесса управления рисками. Он состоит из идентификации опасности, а также анализа и оценки рисков, связанных с влиянием этой опасности;
«опасность» (hazard) - потенциальный источник вреда;
«оценка риска» (risk evaluation) - сравнение предполагаемого риска с установленными критериями приемлемости риска с использованием количественной и качественной шкалы с целью определения значимости риска;
«принятие риска» (risk acceptance) - решение принять риск; «потребность» (requirements) - явные или предполагаемые потребности или ожидания пациентов или лиц, осуществляющих представление их интересов (например, медицинских работников, работников уполномоченных органов);
«риск» (risk) - комбинация вероятности причинения вреда и тяжести этого вреда;
«система качества» (quality system) - совокупность всех аспектов системы, которая внедряет политику в области качества и обеспечивает достижение целей в отношении качества.
«снижение рисков» (risk reduction) - меры, предпринятые для снижения вероятности причинения вреда и серьезности этого вреда;
«способность к выявлению» (detectability) - возможность выявить или установить наличие, присутствие или факт опасности;
«тенденция» (trend) - статистический термин, означающий направление или степень изменения переменной(ых);
«тяжесть» (severity) - мера возможных последствий опасности; «управление рисками» (risk management) - систематическое применение политики управления качеством, процедур и правил с целью общей оценки, контроля, обзора рисков и соответствующего информирования;
«управление рисками для качества» (quality risk management) - систематический процесс общей оценки, контроля, информирования и обзора рисков для качества лекарственного препарата на протяжении его жизненного цикла.
ДОПОЛНЕНИЕ № 1
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к методам и инструментам
управления рисками для качества
Цель настоящего дополнения - представить общий обзор и ссылки на основные инструменты, которые могут быть использованы при управлении рисками для качества (далее - управление рисками) в промышленности и деятельности уполномоченных органов. Эти ссылки приведены с целью расширения знаний и представления более детальной информации относительно конкретного инструмента управления рисками. Данный перечень не является исчерпывающим. Важно отметить, что ни один инструмент или набор инструментов не может быть применим ко всем случаям, когда используется управление рисками.
I.1. Основные вспомогательные методы управления рисками
Некоторыми из простых средств, которые широко применяются для структуризации системы управления рисками путем упорядочения данных и для содействия принятию решений, являются:
блок-схемы;
контрольные карты;
картирование процесса;
диаграммы причин и следствий (называемые также диаграммами Исшикавы или диаграммами «рыбий скелет»).
I.2. Анализ режимов и последствий отказов
(Failure Mode Effects Analysis - FMEA)
FMEA предназначен для оценивания характера потенциальных отказов при проведении процесса, а также возможных последствий отказов для результата процесса и (или) характеристики продукции. Если виды отказов установлены, следует применять снижение рисков с целью устранения, ограничения, уменьшения или контроля потенциальных отказов. FMEA зависит от специфики продукции и процесса. FMEA методически разделяет анализ сложных процессов на стадии, которыми можно управлять. Это мощный инструмент для обобщенного рассмотрения характера важных отказов, факторов, способствующих таким отказам, и возможных последствий таких отказов.
FMEA можно применять в отношении оборудования и помещений, для анализа технологической операции и ее влияния на продукцию или процесс. FMEA определяет элементы (операции) системы, делающие ее уязвимой. Результаты FMEA могут быть использованы в качестве основы для планирования, либо последующего анализа, либо составления рекомендаций относительно использования ресурсов.
I.3. Анализ режимов, последствий и критичности отказов
(Failure Mode,Effects and Criticality Analysis - FMECA)
FMEA может быть расширен, в целях включения также исследования степени тяжести последствий, соответствующей вероятности случаев отказов, способности их выявления. Таким образом, FMEA становится анализом характера, последствий и критичности отказов (Failure Mode, Effects and Criticality Analysis - FMECA). Для проведения такого анализа должны быть установлены спецификации на продукцию и процесс. С помощью FMECA могут быть определены точки, в которых необходимо принятие дополнительных предупреждающих мер, чтобы свести риски к минимуму.
Применять FMECA в фармацевтической промышленности необходимо преимущественно для отказов и рисков, связанных с производственными процессами, однако применение FMECA этим не ограничивается. Результатом FMECA является относительная «шкала» риска для каждого вида отказа, с помощью которой проводят ранжирование режимов на основании относительного риска.
I.4. Анализ древа ошибок (Fault Tree Analysis - FTA)
Анализ древа ошибок (FTA) - подход, предполагающий наличие несоответствий в функциональных характеристиках продукции или процессе. С помощью этого подхода оцениваются одноразовые ошибки системы (или части системы), а также могут быть объединены множественные факторы отказа путем установления причинных связей. Результаты представляют в виде иллюстрации в форме древа видов отказов. На каждом уровне древа комбинации видов отказов могут быть описаны с помощью логических операций («и», «или» и др.). FTA зависит от понимания экспертами процесса в части установления причинных факторов.
FTA можно применять для установления пути к основной причине отказа, а также для расследования претензий или отклонений, позволяющего достичь полного понимания их основных причин и гарантировать, что запланированные улучшения позволят решить проблему и не приведут к возникновению других проблем (то есть решение одной проблемы не должно являться причиной появления другой проблемы). Анализ древа ошибок является эффективным инструментом для оценки степени влияния того, как множественных факторов на данную проблему. Результатом FTA является визуальное выражение видов отказов. FTA полезен как для общего оценивания рисков, так и для разрабатывающихся планов мониторинга.
I.5. Анализ опасностей и критические контрольные точки (Hazard
Analysis and Critical Control Points - HACCP)
HACCP является системным, предупреждающим и профилактическим инструментом для обеспечения качества, надежности и безопасности продукции. Это структурированный подход с применением технических и научных принципов для анализа, оценки, предупреждения и контроля рисков или неблагоприятных последствий опасности, которые являются результатом планирования, разработки, производства и применения продукции.
HACCP состоит из следующих 7 этапов:
проведение анализа безопасности и определение предупреждающих мер для каждой стадии процесса;
определение критических контрольных точек;
установление критических пределов;
установление системы проверки критических контрольных точек;
определение корректирующих мероприятий, которые должны быть проведены, если при мониторинге установлено, что критические контрольные точки являются неконтролируемыми;
введение системы подтверждения эффективности работы системы HACCP;
установление системы хранения записей.
HACCP может быть применен для определения рисков, связанных с физической, химической и биологической опасностью (в том числе с микробной контаминацией), и управления ими. HACCP наиболее полезен, если понимание специфики продукции и процесса является достаточно полным для обеспечения идентификации критических контрольных точек. Результатом HACCP является информация касающаяся управления рисками и облегчающая мониторинг критических точек не только в ходе производственного процесса, но и на других этапах жизненного цикла.
I.6. Анализ опасностей и работоспособности (Hazard Operability
Analysis - HAZOP)
HAZOP основан на теории, допускающей, что случаи рисков являются следствием отклонения от запланированных или рабочих параметров, и является системным методом «мозгового штурма» для идентификации опасности с использованием так называемых «направляющих слов». «Направляющие слова» (например, «нет», «больше», «иной, чем», «часть...» и т. д.) применяются в отношении соответствующих параметров (например, контаминация, температура) для того, чтобы помочь установить возможные отклонения от обычных или запланированных параметров. Часто используется группа людей, обладающих знаниями и опытом по разработке процесса или продукта и его применению.
HAZOP может применяться в отношении производственных процессов (в том числе при контрактном производстве), а также в отношении поставщиков, оборудования и помещений для производства активных фармацевтических субстанций и лекарственных препаратов. HAZOP преимущественно применяется также в фармацевтической промышленности для оценки безопасности процесса. Результатом HAZOP (как и HACCP) является перечень критических операций для управления рисками. Это облегчает регулярный мониторинг критических точек в ходе производственного процесса.
I.7. Предварительный анализ опасности
(Preliminary Hazard Analysis - PHA)
PHA является инструментом, основанным на использовании предыдущего опыта или знаний об опасности или отказе и направленным на определение других факторов опасности, опасных ситуаций и случаев, которые могут быть причиной вреда, а также на оценку вероятности их возникновения в рамках данной деятельности, данных технических средств, продукции или системы.
PHA включает в себя:
идентификацию возможностей того, что произойдет случай, связанный с риском;
качественную оценку масштаба возможного повреждения или вреда для здоровья, которые являются следствием;
относительное ранжирование опасности с использованием комбинации тяжести последствий и вероятности случая; определение возможных корректирующих действий.
PHA может быть полезным при анализе систем или при выявлении первостепенной опасности, если обстоятельства не позволяют применять более масштабный способ. PHA может быть применим при планировании производства продукции, процесса и помещений, а также для оценки типов опасности для основной продукции, для видов продукции и для отдельного продукта. PHA наиболее часто применяется на ранних этапах разработки проекта при недостаточном количестве информации о деталях плана или операционных процедурах. Таким образом, PHA часто является предварительным инструментом для последующих исследований. Как правило, опасность, установленная при применении PHA, в последующем оценивается с помощью других инструментов управления рисками, указанных в настоящем разделе.
I.8. Ранжирование и фильтрация рисков
Ранжирование и фильтрация рисков являются инструментом для сравнения и ранжирования рисков. Ранжирование рисков сложных систем, как правило, требует оценки многочисленных разнообразных количественных и качественных факторов каждого риска. Инструмент заключается в разделении основной связанной с риском проблемы на множество компонентов в целях фиксации факторов, связанных с риском. Эти факторы объединяются в одну относительную шкалу рисков, которую можно применять для ранжирования рисков. Фильтры, которые представляют собой значимые факторы или границы уровней риска, могут быть использованы для градации или ранжирования риска в соответствии с целями управления или политики.
Ранжирование и фильтрацию рисков можно применять при определении приоритетов проведения инспекции (аудита) производственных участков со стороны уполномоченных органов (организаций) государств-членов или со стороны представителей предприятий. Методы ранжирования рисков являются полезными, в частности, в ситуациях, когда риски и последствия, которыми необходимо управлять, являются разнообразными и представляют трудности для сравнения при применении только одного инструмента. Ранжирование рисков целесообразно, если для управления в рамках одной и той же организационной схемы необходимо оценить как количественно, так и качественно оцениваемые риски.
I.9. Статистические методы
Статистические методы могут способствовать управлению рисками для качества и облегчать его осуществление. Они обеспечивают возможность эффективной оценки данных, помогают при определении важности набора(ов) данных, а также способствуют принятию наиболее правильных решений. Перечень некоторых основных статистических методов, широко применяемых в фармацевтической промышленности, включает в себя:
а) контрольные карты, в том числе:
приемочные контрольные карты;
контрольные карты для арифметического среднего с предупреждающими границами;
контрольные карты кумулятивных сумм;
контрольные карты Шухарта;
взвешенное скользящее среднее;
б) планирование экспериментов (Design of Experiments - DOE);
в) гистограммы;
г) диаграммы Парето;
д) анализ возможностей процесса.
ДОПОЛНЕНИЕ № 2
к Правилам надлежащей
производственной практики
Евразийского экономического союзf
Потенциальное применение
управления рисками для качества
Настоящее Дополнение предназначено для определения возможного применения принципов и инструментов управления рисками для качества как представителями промышленности, так и представителями уполномоченных органов государств - членов Евразийского экономического союза (далее соответственно - государства-члены, Союз). Однако выбор инструментов управления рисками для качества полностью зависит от специфических факторов и обстоятельств.
Приведенные примеры представлены в целях иллюстрации и являются только рекомендациями по возможному применению управления рисками для качества.
Настоящее Дополнение не содержит новых требований в дополнение к требованиям, установленным законодательством государств-членов и актами органов Союза.
II. 1. Управление рисками для качества как часть интегрированного управления качеством предполагает:
наличие документации:
для обзора действующих версий и соблюдения требований, законодательства государств-членов и актов органов Союза;для определения необходимости разработки и (или) разработка содержания стандартных операционных процедур (СОПов), руководств и т. п.;
организацию и проведение обучения:
для определения целесообразности начального обучения и (или) постоянных циклов обучения, основывающихся на образовании, опыте и трудовых навыках персонала, а также периодической оценки проведенного обучения (например, его эффективности);
для определения знаний, опыта, квалификационных характеристик и физических возможностей, которые позволяют персоналу выполнять работу правильно и не оказывать отрицательного влияния на качество продукции;
выявление несоответствий качеству:
с целью обеспечения основы для определения и оценки потенциального влияния на качество продукции возможных несоответствий, претензий, тенденций, расследований, результатов, не соответствующих спецификации и т. д., а также информирования о них;
для содействия информированию о риске и определении в сотрудничестве с уполномоченным органом (организацией) государств- членов соответствующего мероприятия (например, отзыв) в связи со значительным дефектом;
планирование аудита (инспекции):
частота и область аудитов, как внутренних, так и внешних, устанавливается с учетом таких факторов, как:
требования законодательства государств-членов и актов органов Союза;
текущее состояние общего соответствия и совокупность имеющихся сведений о предприятии или производственной площадке;
надежность деятельности организации в плане управления рисками для качества; сложность участка;
сложность производственного процесса; сложность продукции и ее терапевтическое значение; количество и значимость дефектов (например, количество ведущих к отзывам продукции);
результаты предыдущих аудитов (инспекций); существенные изменения помещений, оборудования, процессов, ключевого персонала;
опыт производства продукции (например, частота производства, объем и количество серий);
результаты испытаний в аккредитованных испытательных лабораториях;
осуществление периодического обзора:
для выбора, оценки и объяснения данных, которые свидетельствуют о тенденции в рамках обзора качества продукции;
для объяснения данных мониторинга (например, для систематической оценки надлежащего проведения повторной валидации или изменений при отборе проб);
управление изменениями (контроль изменений): для управления изменениями на основании знаний и информации, полученной во время фармацевтической разработки и производства;
для оценки влияния изменений на соответствие качества готовой продукции требованиям спецификации;
для оценки влияния на качество продукции изменений, внесенных в производственные площадки, оборудование, материалы, производственный процесс, или переносов технологии;
для определения мероприятий, которые предшествуют внесению изменения, например, дополнительные испытания, квалификация (повторная квалификация), валидация (повторная валидация) или информирование уполномоченных органов (организаций) государств- членов;
постоянное улучшение системы управления рисками: для содействия постоянному улучшению процессов на протяжении жизненного цикла продукции.
II.2. Управление рисками для качества как часть деятельности уполномоченных органов (организаций) государств-членов предполагает проведение следующих мероприятий:
инспекция и систематическая оценка деятельности: для содействия рациональному распределению ресурсов, в том числе, например, для планирования инспекций и частоты их проведения, а также для проведения инспекций и определения их объема (см. «Планирование аудита (инспекции) » в пункте 1 настоящего дополнения);
для оценки значимости результатов (например, дефектов, ведущих к отзывам продукции) и данных, полученных при инспектировании;
для определения необходимости и вида предусмотренных законодательством мероприятий по результатам проведения инспекции;
для оценки информации, предоставленной представителями предприятий, в том числе относительно фармацевтической разработки; для оценки влияния предлагаемых отклонений или изменений; для определения рисков, которые следует обсуждать с инспекторами и экспертами для содействия лучшему пониманию того, как риск можно контролировать или как он контролируется (например,
выпуск по параметрам, процессно-аналитическая технология (Process Analytical Technology - PAT)).
II.3. Управление рисками для качества как часть разработки предполагает проведение следующих мероприятий:
для планирования качества продукции и технологического процесса, в целях получения продукции с функциональными характеристиками, соответствующими ее назначению;
для расширения знаний о функциональных характеристиках продукции в зависимости от изменения характеристик сырья в широком диапазоне (например, распределение частиц по размерам, содержание влаги, реологические свойства), изменение технологических операций и параметров процесса;
для оценки критических характеристик исходного сырья, растворителей, исходного сырья для активных фармацевтических субстанций (АФС), самих АФС, вспомогательных веществ или упаковочных материалов;
для установления соответствующих спецификаций, определения критических параметров процесса и организации производственного контроля (например, на основании информации о клинически значимых показателях качества, полученной на этапе фармацевтической разработки, а также о возможности контролировать их в ходе процесса);
для уменьшения непостоянства показателей качества (снижение количества дефектов продукции, исходного сырья и материалов, отклонений при производстве);
для оценки необходимости проведения дополнительных исследований (например, биоэквивалентность, стабильность) при масштабировании и переносе технологии;
для использования концепции «пространства проектных параметров».
II.4. Управление рисками для качества в отношении производственных помещений, оборудования и систем обеспечения предполагает проведение следующих мероприятий:
проектирование производственных помещений и (или) оборудования:
для определения соответствующих зон при проектировании зданий и производственных помещений, включая в том числе: определение направлений потоков материалов и персонала; сведение к минимуму контаминации; проведение мероприятий по контролю паразитов; предупреждение перепутывания;
сравнение оборудования открытого и закрытого типов; сравнение чистых помещений с изолирующей технологией; определение специально предназначенных или выделенных производственных помещений и (или) оборудования;
для определения соответствующих материалов для оборудования и контейнеров, контактирующих с продукцией (например, выбор марки нержавеющей стали, сальников, смазочных материалов);
для определения соответствующих систем обеспечения (например, пар, газы, источник питания, сжатый воздух, система нагрева, вентиляции и кондиционирования воздуха, вода);
для определения профилактического обслуживания связанного друг с другом оборудования (например, перечень необходимых запасных частей);
в рамках гигиены в помещениях:
для защиты продукции от опасности со стороны производственной среды, в том числе от химических, микробиологических и физических факторов опасности (например, определение надлежащей одежды и организация гардеробной, гигиены);
для защиты производственной среды и персонала от опасностей, связанных с производимой продукцией вследствие перекрестной контаминации;
в рамках квалификации производственных помещений (оборудования, систем обеспечения) - определение области и масштаба квалификации помещений, зданий и технологического оборудования, а также лабораторных приборов (в том числе надлежащих методов калибровки);
в рамках очистки оборудования и контроль производственной среды:
для рационального распределения усилий и принятия решений, с учетом предназначения оборудования (например, многоцелевое или специально предназначенное оборудование, серийное производство или непрерывный технологический процесс);
для определения пределов приемлемости для валидации очистки; в рамках проведения калибровка (профилактического обслуживания) - составление графиков калибровки и профилактического обслуживания;
в рамках использования компьютерных систем и оборудования, контролируемого с помощью компьютеров:
для выбора конфигурации компьютеров и программного обеспечения (например, модульная, структурированная, устойчивая к сбоям система);
для определения масштаба валидации, в том числе:
определение критических функциональных параметров; выбор требований и конструкции; проверка кодов;
масштаб испытаний и методы испытаний; правильность электронных записей и подписей.
II.5. Управление рисками для качества как часть управления исходным сырьем и материалами предполагает проведение следующих мероприятий:
определение и оценка поставщиков и производителей по контракту для обеспечения всесторонней оценки поставщиков и производителей по контракту (например, проведение их аудита, заключение соглашений с поставщиками относительно качества):
контроль исходного сырья для оценки различий и возможных рисков для качества, связанных с изменчивостью исходного сырья (например, срок хранения, схема синтеза);
в рамках использования исходного сырья и материалов: для определения того, являются ли приемлемыми для использования исходное сырье и материалы, находящиеся в карантине (например, для дальнейшего технологического процесса);
для определения надлежащего осуществления повторной обработки, переработки, использования возвращенной продукции;
в рамках обеспечения условий хранения и оптовой торговли, логистики:
для оценки полноты соглашений по обеспечению соответствующих условий хранения и транспортирования (например, температура, влажность, конструкция контейнера);
для определения влияния несоответствий в условиях хранения и транспортирования (например, «обеспечение холодовой цепи»);
для функционирования инфраструктуры (например, возможность обеспечивать надлежащие условия отгрузки, временного хранения, обращение с опасными материалами и подконтрольными веществами, таможенная очистка);
для предоставления информации об обеспечении пригодности лекарственных препаратов (например, ранжирование рисков для цепи поставки).
II.6. Управление рисками для качества, как часть производства предполагает проведение следующих мероприятий:
валидация:
для определения области и масштаба осуществления деятельности по подтверждению, квалификации и валидации (например, аналитические методики, процессы, оборудование и процедуры очистки);
для определения масштаба осуществления последующих действий (например, отбор проб, мониторинг и повторная валидация);
для разграничения критических и некритических стадий процесса для помощи в планировании валидационных испытаний;
отбор проб (испытание в ходе процесса производства): для оценки частоты и масштаба проведения контрольных испытаний в процессе производства (например, для обоснования уменьшения объема испытаний в условиях доказанного контроля);
для оценки и обоснования использования процессно-аналитической технологии (PAT) наряду с выпуском по параметрам и выпуском в реальном времени;
планирование производства - составление соответствующего плана производства (например, специально предназначенное производство, производство кампаниями (циклами производства) и очередность сопутствующих технологических процессов).
II.7. Управление рисками для качества как часть лабораторного контроля и исследований стабильности предполагает проведение следующих мероприятий:
в рамках результатов, не соответствующих спецификация - установление возможных основных причин и определение корректирующих мероприятий в ходе расследования результатов, не соответствующих спецификациям;
в рамках определения периода до проведения повторных испытаний (даты окончания срока годности) - оценка правильности хранения и проведение испытаний промежуточной продукции, вспомогательных веществ и исходного сырья.
II.8. Управление рисками для качества как часть упаковки и маркировки предполагает проведение следующих мероприятий:
проектирование вторичной упаковки, предназначенной для защиты первичной упаковки продукции (например, чтобы обеспечить подлинность продукции, разборчивую надпись на этикетке);
определение критических характеристик системы укупорки контейнера при выборе системы укупорки контейнера;
планирование процедур контроля этикеток, с учетом возможности перепутывания этикеток различной продукции, в том числе разных версий одной и той же этикетки.
Глава III. Фармацевтическая система качества
Предисловие
Производителям лекарственных средств следует разработать и внедрить эффективную фармацевтическую систему качества, чтобы обеспечить соответствие требованиям настоящих Правил и придерживаться положений, изложенных в главе 1 части I настоящих Правил.
Настоящая глава описывает подход к фармацевтической системе качества, которую можно применять на различных стадиях жизненного цикла продукции. Следовательно, указания данной главы выходят за рамки требований настоящих Правил (поскольку настоящие Правила, за исключением производства лекарственных средств для клинических исследований, не распространяются на часть жизненного цикла лекарственного средства, которая связана с его разработкой). Данный раздел дополняет требования настоящих Правил, позволяет внедрить на предприятии фармацевтическую систему качества. Использование положений настоящей главы будет способствовать инновациям, постоянному улучшению и усилению взаимосвязи между фармацевтической разработкой и производственной деятельностью.
1. Фармацевтическая система качества
1.1. Введение
Настоящая глава гармонизирована с Руководством ICH Q10, которое описывает модель эффективной системы управления качеством для фармацевтической промышленности (далее - фармацевтическая система качества). В контексте настоящей главы термин «фармацевтическая система качества» относится к модели, описанной в документе ICH Q10.
В документе ICH Q10 описана единая всесторонняя модель эффективной фармацевтической системы качества, которая основывается на концепции качества Международной организации по стандартизации (ISO), включает соответствующие положения правил надлежащего производства и дополняет документы ICH Q8 «Фармацевтическая разработка» и ICH Q9 «Управление рисками для качества». Настоящая глава является моделью для фармацевтической системы качества, которая может применяться на различных этапах жизненного цикла продукции. Большинство положений настоящей главы, применяемых к производственным предприятиям, подробно излагается в других разделах настоящих Правил. Настоящая глава предназначена для установления каких-либо новых положений, которые выходят за рамки действующих нормативных требований. Положения, содержащиеся в настоящей главе, дополняют настоящие Правила и не являются обязательными.
Настоящая глава служит доказательством того, что производители и уполномоченные органы поддерживают эффективную фармацевтическую систему качества с целью улучшения качества и доступности лекарственных препаратов во всем мире в интересах здравоохранения. Применение настоящей главы на протяжении жизненного цикла продукции будет способствовать инновациям и постоянному улучшению, а также упрочнению связи между фармацевтической разработкой и производственной деятельностью.
1.2. Область применения
Настоящая глава распространяется на системы, обеспечивающие фармацевтическую разработку и производство активных фармацевтических субстанций (АФС), а также лекарственных препаратов, включая лекарственные биотехнологические и биологические лекарственные препараты, на протяжении всех этапов жизненного цикла продукции.
Структурные элементы настоящей главы должны применяться соответственно и пропорционально каждому этапу жизненного цикла продукции с учетом различий между этими этапами и различной цели каждого этапа (см. раздел 3 настоящей главы).
Для целей настоящего документа жизненный цикл продукции включает следующую производственную деятельность новых и уже существующих лекарственных препаратов: на этапе фармацевтической разработки: разработка активной фармацевтической субстанции; разработка состава (включая систему «контейнер (средство укупорки»));
производство лекарственных препаратов для клинических исследований;
разработка системы доставки (при необходимости);
разработка технологического процесса и масштабирование; разработка методик анализа;
на этапе переноса технологии:
перенос новой продукции из разработки в производство;
перенос технологии производства зарегистрированной продукции внутри или между производственными или контролирующими подразделениями;
на этапе промышленного производства:
приобретение и контроль исходного сырья и материалов;
предоставление помещений для производства, систем обеспечения и оборудования;
технологический процесс (включая упаковку и маркировку);
контроль качества и обеспечение качества;
выдача разрешения на выпуск продукции;
хранение;
реализация (за исключением оптовой торговли);
на этапе прекращения выпуска продукции:
хранение документации;
хранение образцов;
продолжающаяся оценка продукции и составление отчетов.
1.3. Взаимосвязь настоящей главы с требованиями
части I настоящих Правил, стандартами ISO и документом ICH Q7
Требования настоящих Правил, Руководство 1СН Q7 «Правила надлежащего производства активных фармацевтических субстанций» и Руководства системой управления качества ISO являются основой настоящей главы. Для достижения изложенных ниже целей настоящая глава расширяет часть I настоящих Правил описанием характерных элементов системы качества и ответственности руководства. Настоящий раздел обеспечивает модель, сочетающую фармацевтическую систему качества на всех этапах жизненного цикла продукции с требованиями настоящих Правил для их совместного применения.
Требования настоящих Правил не направлены непосредственно на все этапы жизненного цикла продукции (например, на разработку). Элементы фармацевтической системы качества и ответственность руководства, описанные в настоящей главе, предназначены для содействия применению научных подходов и подходов, основанных на оценке рисков, на каждом этапе жизненного цикла продукции, способствуя тем самым постоянному улучшению продукции на протяжении всего жизненного цикла.
1.4. Взаимосвязь настоящего документа с принципами
государственного контроля
Принципы государственного контроля в отношении определенной продукции или предприятия-производителя должны соответствовать уровню продукции, пониманию процесса, результатам управления рисками для качества и эффективности фармацевтической системы качества. Эффективность фармацевтической системы качества после ее внедрения может подтверждаться в обычном режиме при проведении инспекций предприятия уполномоченными органами. Потенциальные возможности улучшения научных и основанных на анализе рисков надзорных подходов изложены в дополнении № 1 к настоящим Правилам. Принципы государственного контроля определяются законодательством соответствующего государства-члена.
1.5. Цели настоящего документа
Внедрение модели фармацевтической системы качества должно привести к достижению 3 основных целей, которые дополняют или совершенствуют требования настоящих Правил:
достичь реализации продукции (создать, внедрить и поддерживать систему, которая обеспечивает поставку продукции с показателями качества, соответствующими потребностям пациентов, медицинских работников, уполномоченных органов (включая соответствие установленным требованиям), а также внутренних и внешних потребителей);
установить и поддерживать контролируемое состояние (разработать и использовать эффективные системы мониторинга и контроля эффективности процесса и качества продукции, обеспечивая таким образом гарантию пригодности и возможностей процессов. При организации систем мониторинга и контроля может быть полезным управление рисками для качества);
способствовать постоянному улучшению (выявлять и внедрять соответствующие методы улучшения качества продукции и процессов, снижения их нестабильности принимать инновационные решения и улучшать фармацевтическую систему качества, обеспечивая таким образом постоянное удовлетворение потребностей в отношении качества. При выявлении приоритетных областей, требующих постоянного улучшения, может быть полезным управление рисками для качества).
1.6. Средства улучшения: управление знаниями и управление
рисками для качества
Управление знаниями и управление рисками для качества являются средствами, которые дают возможность предприятиям эффективно и успешно применить настоящую главу. Эти средства будут способствовать достижению целей, описанных в пункте 1.5настоящей главы, и обеспечат основания для принятия решений, основанных на научных знаниях и знаниях о рисках в отношении качества продукции.
1.6.1. Управление знаниями
Знаниями о продукции и процессах необходимо управлять при разработке, на протяжении периода нахождения продукции на рынке и до прекращения ее производства и медицинского применения. Например, деятельность при разработке с использованием научных подходов обеспечивает знание продукции и понимание процессов. Управление знаниями является систематическим подходом, который заключается в приобретении, анализе, накоплении и распространении информации о продукции, технологических процессах и компонентах. Источники информации включают в себя, в том числе, первично знания (общеизвестные или документально оформленные внутри предприятия), исследования в области фармацевтических разработок, деятельность по переносу технологий, исследования по валидации процессов на протяжении жизненного цикла продукции, опыт производства, инновации, постоянное улучшение и деятельность, направленную на управление изменениями.
1.6.2. Управление рисками для качества
Управление рисками для качества является неотъемлемой частью фармацевтической системы качества. Оно может обеспечить профилактический подход к выявлению, научной оценке и контролю потенциальных рисков для качества. Это способствует постоянному улучшению эффективности процесса и качества продукции на всех этапах жизненного цикла. Положения главы IIнастоящей части Правил представляют принципы и примеры инструментов по управлению рисками для качества, которые могут применяться к различным аспектам фармацевтического качества.
1.7. Структура и содержание фармацевтической системы качества
Структура, организация и документальное оформление фармацевтической системы качества должны быть понятными и четко структурированными для облегчения общего понимания и последовательного применения.
При разработке новой фармацевтической системы качества или при модификации существующей системы необходимо учитывать объем и сложность деятельности предприятия. В структуру фармацевтической системы качества должны быть включены соответствующие принципы управления рисками. Хотя некоторые аспекты фармацевтической системы качества могут применяться к деятельности всего предприятия, а иные - только к определенным подразделениям, эффективность внедрения фармацевтической системы качества обычно демонстрируется на уровне подразделения.
Фармацевтическая система качества должна включать соответствующие процессы, ресурсы и ответственность для обеспечения качества работ по контракту и закупаемого сырья и материалов, как описано в пункте 2.7 настоящей главы.
В рамках фармацевтической системы качества должна определяться ответственность руководства, как описано в разделе 2 настоящей главы.
Фармацевтическая система качества должна включать следующие элементы: мониторинг эффективности процесса и качества продукции, корректирующие и предупреждающие действия, управление изменениями и проверки со стороны руководства, как описано в разделе 3 настоящей главы.
Основные показатели результативности должны выявляться и использоваться для проверки эффективности процессов в рамках фармацевтической системы качества, как описано в разделе 4 настоящей главы.
1.8. Руководство по качеству
Должно быть разработано руководство по качеству или равноценный документ, включающий в себя описание фармацевтической системы качества, в том числе:
политику в области качества;
описание сферы применения фармацевтической системы качества;
определение процессов в рамках фармацевтической системы качества, а также их последовательности, взаимосвязи и взаимозависимости. Карты процессов и графики потоков могут быть полезными инструментами для облегчения описания их в графическом виде;
описание ответственности руководства в рамках фармацевтической системы качества.
2. Ответственность руководства
Руководство предприятия несет ответственность за установление и обеспечение выполнения обязательств в отношении качества продукции и эффективности фармацевтической системы качества на уровне предприятия.
2.1. Обязанности руководства
а) При достижении целей в сфере качества высшее руководство несет основную ответственность за обеспечение внедрения эффективной фармацевтической системы качества, определение обязанностей, ответственности и полномочий, доведение их до сведения персонала, а также исполнение их всеми подразделениями предприятия.
b) Руководство должно:
1) участвовать в разработке, внедрении, мониторинге и поддержании фармацевтической системы качества;
2) демонстрировать поддержку фармацевтической системы качества и гарантировать ее внедрение на всех уровнях предприятия;
3) обеспечивать наличие своевременных и эффективных средств связи и процедур распространения информации для доведения сведений о проблемах качества до руководства соответствующего уровня;
4) определять персональные и коллективные обязанности, ответственность и полномочия, а также взаимосвязи всех структурных подразделений, имеющих отношение к фармацевтической системе качества. Гарантировать, что эти взаимосвязи установлены и поняты на всех уровнях организации. Требуется наличие независимого отдела (подразделения), ответственного за качество, имеющего полномочия на осуществление определенных обязанностей, связанных с фармацевтической системой качества;
5) проводить проверки в отношении эффективности процесса и качества продукции, а также фармацевтической системы качества;
6) поддерживать постоянное улучшение;
7) выделять соответствующие ресурсы.
2.2. Политика в области качества
а) Руководство должно установить политику в области качества, которая описывает общие цели и направления деятельности предприятия в отношении качества.
b) Политика в области качества должна включать в себя цели выполнения установленных требований, а также способствовать постоянному улучшению фармацевтической системы качества.
c) Политика в области качества должна доводиться до сведения и разъясняться персоналу на всех уровнях предприятия.
d) Для обеспечения постоянной эффективности политика в области качества должна периодически пересматриваться.
2.3. Планирование качества
a) Высшее руководство должно обеспечивать определение и доведение до сведения персонала целей качества, необходимых для внедрения политики в области качества.
b) Цели качества должны поддерживаться на всех уровнях предприятия.
c) Цели качества должны соответствовать стратегическим планам предприятия и согласовываться с политикой в области качества.
d) Руководство должно предоставлять соответствующие ресурсы и возможность обучения для достижения целей качества.
e) Следует устанавливать, контролировать, постоянно доводить до сведения и выполнять показатели эффективности, которые служат мерой достижения целей качества, в соответствии с пунктом 4.1 настоящей главы.
2.4. Управление ресурсами
a) Руководство должно определять и предоставлять достаточные ресурсы (человеческие, финансовые, материальные, помещения и оборудование) для внедрения и поддержания фармацевтической системы качества и постоянного улучшения ее эффективности.
b) Руководство должно обеспечивать надлежащее использование ресурсов соответственно продукции, процессу или производственной площадке.
2.5. Внутренний обмен информацией
a) Руководство должно обеспечить установление и внедрение внутри предприятия механизмов соответствующего обмена информацией.
b) Процессы обмена информацией должны обеспечивать передачу потока соответствующей информации между всеми уровнями предприятия.
c) Процессы обмена информацией должны обеспечивать своевременную передачу информации об определенных проблемах с качеством продукции и с фармацевтической системой качества.
2.6. Проверки, проводимые руководством
a) Высшее руководство посредством проведения проверок должно нести ответственность за управление фармацевтической системой качества для обеспечения ее эффективности и постоянного соответствия установленным требованиям.
b) Руководство должно оценивать результаты периодических проверок эффективности процесса и качества продукции, а также фармацевтической системы качества, в соответствии с разделами 3 и 4 настоящей главы.
2.7. Управление работами по контракту и
закупаемыми материалами
Фармацевтическая система качества, включая описанную в настоящем разделе ответственность руководства, распространяется на контроль и проверку работ по контракту и качество закупаемого исходного сырья и материалов. Производитель лекарственных средств несет, в конечном счете, ответственность за гарантию осуществления процессов, необходимых для обеспечения контроля работ по контракту и контроля качества закупаемых исходного сырья и материалов. Эти процессы должны быть объединены с управлением рисками для качества и должны включать в себя:
a) осуществление оценки (например, путем проведения аудитов, контроля исходного сырья и материалов, проведения квалификации) исполнителей работ по контракту и поставщиков сырья. Такая оценка осуществляется до проведения работ по контракту или до выбора поставщиков исходного сырья и материалов на предмет их пригодности и соответствия требованиям к выполнению таких работ или поставки исходного сырья и материалов с использованием установленной цепи поставки;
b) определение сфер ответственности и процессов передачи информации в отношении работ, связанных с качеством, в которые вовлечены заинтересованные стороны. В отношении работ по контракту это должно быть включено в письменное соглашение между заказчиком и исполнителем;
c) мониторинг и проверка деятельности исполнителя или качества исходного сырья и материалов от поставщика, а также определение и внедрение необходимых улучшений;
d) мониторинг исходного сырья и материалов для обеспечения гарантии того, что они получены из утвержденных источников с использованием согласованной цепи поставки.
2.8. Управление изменениями в праве
собственности на продукцию
В случае если происходит изменение в праве собственности на продукцию (например, в результате покупки), руководство должно принимать во внимание сложность этого процесса и гарантировать следующее:
a) для каждой задействованной в процессе стороны определена текущая ответственность;
b) передана вся необходимая информация.
3. Постоянное улучшение эффективности процесса и
качества продукции
В этом разделе описаны цели фармацевтической системы качества на различных этапах жизненного цикла продукции, а также 4 характерных элемента фармацевтической системы качества, которые расширяют требования настоящих Правил для достижения целей данного документа, определенных в разделе 1.5 настоящей главы. Положения настоящего раздела не изменяют требования настоящих Правил.
3.1. Цели на этапах жизненного цикла продукции
Ниже описаны цели для каждого этапа жизненного цикла продукции.
3.1.1. Фармацевтическая разработка
Целью деятельности по фармацевтической разработке является разработка продукции и процесса ее производства для постоянного обеспечения ожидаемых характеристик и удовлетворения потребностей пациентов, медицинских работников, требований уполномоченных органов и внутренних потребителей. Подходы к фармацевтической разработке описаны в соответствующих нормативных правовых актах. Результаты исследовательских работ и клинических исследований, которые не входят в сферу применения настоящего документа, являются исходными данными для фармацевтической разработки.
3.1.2. Перенос технологии
Целью деятельности, связанной с переносом технологии, является передача знаний о продукции и процессе от разработчиков к производителям, а также внутри или между производственными площадками для производства продукции, соответствующей своему предназначению. Эти знания составляют основу процесса производства, стратегии контроля, подхода к процессу валидации и непрерывного улучшения.
3.1.3. Промышленное производство
Целями промышленного производства являются производство продукции, соответствующей своему предназначению, установление и поддержание контролируемого состояния и способствование постоянному улучшению. Фармацевтическая система качества должна обеспечивать постоянное поддержание желаемого качества продукции, достижение необходимой эффективности процесса, соответствие различных средств контроля, определение и оценку возможностей улучшения, а также постоянное увеличение объема знаний.
3.1.4. Прекращение производства продукции
Целью проведения мероприятий по прекращению производства продукции является эффективное управление конечным этапом жизненного цикла продукции. Для управления мероприятиями по прекращению производства продукции должны использоваться предварительно установленные подходы: хранение документации и образцов, постоянная оценка продукции (например, работа с претензиями и испытания стабильности), составление отчетов в соответствии с установленными требованиями.
3.2. Элементы фармацевтической системы качества
Элементы, описанные ниже, частично входят в другие разделы настоящих Правил и приложения к настоящим Правилам. Такая модель предназначена для усовершенствования этих элементов с целью содействия применению подхода к качеству, основанного на жизненном цикле продукции. Это следующие четыре элемента:
система мониторинга эффективности процесса и качества продукции; система корректирующих и предупреждающих действий (CAPA); система управления изменениями;
проверка со стороны руководства эффективности процесса и качества продукции.
Эти элементы должны применяться соответственно и пропорционально каждому из этапов жизненного цикла продукции с учетом различий между ними и различной цели каждого этапа. На каждом этапе жизненного цикла продукции предприятия должны оценивать перспективы для внедрения инновационных подходов по улучшению качества продукции.
Описание каждого элемента завершается таблицей с примерами применения каждого элемента к конкретному этапу жизненного цикла продукции.
3.2.1. Система мониторинга эффективности процесса и качества
продукции
Производители лекарственных средств должны планировать и применять систему мониторинга эффективности процесса и качества продукции, а также обеспечивать поддержание контролируемого состояния. Эффективность системы мониторинга предусматривает обеспечение постоянной производительности процессов и средств контроля для производства продукции желаемого качества и выявления сфер для постоянного улучшения.
Система мониторинга эффективности процесса и качества продукции должна:
a) использовать управление рисками для качества для установления стратегии контроля. Такая стратегия может включать параметры и характеристики, относящиеся к активным фармацевтическим субстанциям, исходному сырью и компонентам лекарственных препаратов, условиям эксплуатации помещений и оборудования, контролю в процессе производства, спецификациям на готовую продукцию, а также связанные методы и частоту проведения мониторинга и контроля. Стратегия контроля должна способствовать своевременной обратной (прямой) связи и соответствующим корректирующим и предупреждающим действиям;
b) обеспечивать инструменты для измерения и анализа параметров и свойств, определенных в стратегии контроля (например, управление данными и статистические инструменты);
c) анализировать параметры и характеристики, определенные в стратегии контроля, для подтверждения постоянной работы в контролируемом состоянии;
d) определить источники отклонений, которые влияют на эффективность процесса и качество продукции, с целью возможного постоянного улучшения, направленного на снижение или контроль изменчивости;
e) содержать информацию о качестве продукции, поступившую в виде обратной связи как от внутренних, так и от внешних источников, например, претензии, отказы от продукции, несоответствия, отзывы продукции, отклонения, аудиторские проверки и инспекции уполномоченных органов и их заключения;
f) обеспечивать знаниями для улучшения понимания процесса, расширять пространство проектных параметров (если они установлены) и давать возможность применения инновационных подходов к валидации процесса.
Таблица 1
Применение системы мониторинга эффективности процесса и
качества продукции на протяжении жизненного цикла продукции
|
Фармацевтическая разработка |
Перенос технологии |
Промышленное производство |
Прекращение производства продукции |
|
Знания о процессе и продукции и результаты их мониторинга, полученные в процессе разработки лекарственного препарата, можно использовать для установления стратегии контроля на этапе производства |
Мониторинг при проведении работ по масштабированию может обеспечить предварительную оценку эффективности процесса и успешное внедрение в производство. Знания, полученные во время переноса технологий и масштабирования, могут быть полезными для дальнейшей разработки стратегии контроля |
Четко определенная система мониторинга эффективности процесса и качества продукции должна применяться для обеспечения эффективности в контролируемом состоянии и для выявления сфер, требующих улучшения |
При прекращении производства мониторинг (например, проведение исследования стабильности) должен продолжаться до завершения всех испытаний (испытаний). В соответствии с установленными требованиями испытания необходимо продолжать проводить на лекарственных препаратах, находящихся в обороте |
3.2.2. Система корректирующих и предупреждающих действий
Производитель лекарственных средств должен иметь систему корректирующих и предупреждающих действий, которые являются результатом расследования претензий, отказов от продукции, несоответствий, отзывов, отклонений, аудиторских заключений и инспекций уполномоченных органов, а также тенденций, выявленных при проведении мониторинга эффективности процесса и качества продукции. С целью выявления основной причины в процессе расследования должен использоваться структурированный подход. Уровень усилий, формализации и документального оформления расследования должен быть соразмерным уровню рисков согласно главе II настоящей части Правил. Результатом методологии корректирующих и предупреждающих действий должно быть улучшение продукции и процесса, а также улучшение понимания продукции и процесса.
Таблица 2
Применение системы корректирующих и предупреждающих действий на
протяжении жизненного цикла продукции
|
Фармацевтическая разработка |
Перенос технологии |
Промышленное производство |
Прекращение производства продукции |
|
Исследуется изменчивость продукции или процесса. Методология CAPA может быть полезной, когда корректирующие и предупреждающие действия внедрены в итеративную схему и процесс разработки |
CAPA может использоваться в качестве эффективной системы обратной связи, прямой связи и постоянного улучшения |
Следует использовать систему CAPA, при этом должна оцениваться эффективность предпринятых действий |
Система CAPA должна продолжать применяться после прекращения производства продукции. Должно учитываться влияние на продукцию, которая остается в обороте, а также на другую продукцию, на которую могло быть оказано влияние |
3.2.3. Система управления изменениями
Инновации, постоянное улучшение, результаты мониторинга эффективности процесса и качества продукции и система CAPA (корректирующие и предупреждающие действия, corrective action and preventive action, CAPA корректирующие и действия) приводят к изменениям. Для правильной оценки, утверждения и внедрения этих изменений предприятие должно иметь эффективную систему управления изменениями.
В общем случае существуют различия в соблюдении формальной стороны процессов управления изменениями до первоначальной подачи документов в уполномоченный орган и после такой подачи, если изменения к поданным документам могут требоваться в соответствии с установленными нормами.
Система управления изменениями обеспечивает осуществление постоянного, своевременного и эффективного улучшения. Она должна обеспечивать высокую степень уверенности в том, что изменение не приведет к непредсказуемым последствиям.
Система управления изменениями в зависимости от этапов жизненного цикла продукции включает в себя следующее:
a) управление рисками для качества, которое должно использоваться для оценки предлагаемых изменений. Предпринимаемые действия и соблюдение формальностей при оценке должны соответствовать уровню рисков;
b) оценка предлагаемых изменений в отношении регистрационного досье, включая пространство проектных параметров (если они установлены) и (или) текущее понимание продукции и процессов. Оценка должна быть проведена для определения того, требуется ли внесение изменений в регистрационное досье в соответствии с установленными нормами. Работа внутри проектных параметров не считается изменением с точки зрения нормативных требований. Однако, с точки зрения фармацевтической системы качества, все изменения должны оцениваться с помощью системы управления изменениями на предприятии;
c) оценка предложенных изменений группой экспертов, которые имеют необходимый опыт и знания в соответствующей области (например, в сфере фармацевтической разработки, производства, качества, медицины, взаимодействия с уполномоченными органами). Должно гарантироваться, что изменения обоснованы с технической точки зрения. Для предлагаемого изменения следует установить будущие критерии оценки;
d) оценка проведенных изменений после их внедрения для подтверждения того, что цель изменений была достигнута и что это не отразилось негативно на качестве продукции.
Таблица 3
Применение системы управления изменениями на протяжении
жизненного цикла продукции
|
Фармацевтическая разработка |
Перенос технологии |
Промышленное производство |
Прекращение производства продукции |
|
Изменение является неотъемлемой частью процесса разработки и должно быть оформлено документально. Степень формальности процесса управления изменениями должна соответствовать стадии фармацевтической разработки |
Система управления изменениями должна обеспечить управление и документальное оформление поправок, внесенных в процесс при переносе технологии |
При промышленном производстве должна быть внедрена официальная система управления изменениями. Контроль, осуществляемый отделом качества, должен обеспечивать соответствующие оценки, основанные на научных знаниях и знаниях по анализу рисков |
Любые изменения после прекращения производства продукции должны проходить через соответствующую систему управления изменениями |
3.2.4. Проверка руководством эффективности процесса и качества
продукции
Проверка, проводимая руководством, должна гарантировать управление эффективностью процесса и качеством продукции на протяжении жизненного цикла продукции. В зависимости от размера и сложности структуры предприятия, проверка со стороны руководства может состоять из серии проверок на различных уровнях управления. Она должна включать своевременный и эффективный обмен информацией, а также механизмы доведения до сведения высшего руководства информации о соответствующих проблемах, связанных с качеством, для их оценки.
a) Система проверок, проводимых руководством, должна включать в себя:
1) результаты инспекций уполномоченных органов и их заключения, аудиторские и другие оценки, обязательства перед уполномоченными органами;
2) периодические обзоры качества продукции, в том числе:
i) оценку степени удовлетворенности потребителей посредством расследования претензий и анализа отзывов продукции;
ii) заключения по результатам мониторинга эффективности процесса и качества продукции;
iii) эффективность изменений процесса и продукции (включая изменения, которые являются результатом корректирующих и предупреждающих действий);
3) любые последующие мероприятия после проверок, проводимых руководством.
b) Система проверок, проводимых руководством, должна определять соответствующие мероприятия, например:
1) улучшение процесса производства и продукции;
2) обеспечение ресурсами, организация обучения и (или) перераспределение ресурсов;
3) приобретение и распространение знаний.
Таблица 4
Проведение руководством проверок эффективности процесса и
качества продукции на протяжении жизненного цикла продукции
|
Фармацевтическая разработка |
Перенос технологии |
Промышленное производство |
Прекращение производства продукции |
|
Подходы, основанные на проверках, проводимых руководством, могут применяться для обеспечения соответствия разработки продукции и технологического процесса |
Подходы, основанные на проверках, проводимых руководством, должны применяться для обеспечения возможности производства разработанной продукции в промышленном масштабе |
Проверка, проводимая руководством, должна быть структурированной системой, как это изложено выше, и должна содействовать постоянному улучшению |
Проверка, проводимая руководством, должна включать в себя, стабильность продукции и расследование претензий в отношении ее качества |
4. Постоянное улучшение фармацевтической системы качества
В этом разделе описаны мероприятия, которые необходимо проводить для управления и постоянного улучшения фармацевтической системы качества.
4.1. Проверка руководством фармацевтической системы качества
Руководство должно использовать формализованные процессы для проведения периодических проверок фармацевтической системы качества. Такие проверки включают в себя:
а) Систему мер для достижения целей, связанных с фармацевтической системой качества;
b) Оценку показателей эффективности, которые могут использоваться для проверки эффективности следующих процессов в рамках фармацевтической системы качества:
1) претензии, отклонения, САРА и процессы управления изменениями;
2) обратная связь в отношении работ по контракту;
3) процессы самооценки, включая оценку рисков, анализ тенденций и аудиты;
4) внешние оценки, в том числе инспекции уполномоченных органов и их заключения, а также аудиты потребителей.
4.2. Мониторинг внутренних и внешних факторов, которые влияют
на фармацевтическую систему качества
Факторы, проверяемые руководством, могут включать в себя:
a) новые нормативные требования, руководства и публикации относительно качества, которые могут влиять на фармацевтическую систему качества;
b) инновации, способные улучшить фармацевтическую систему качества;
c) изменения условий и целей деятельности;
d) изменения в праве собственности на продукцию.
4.3. Результаты проверки, проводимой руководством, и мониторинга
Итоги проверки руководством фармацевтической системы качества и мониторинга внутренних и внешних факторов могут включать в себя:
а) улучшение фармацевтической системы качества и связанных процессов;
b) распределение или перераспределение ресурсов и (или) обучение персонала;
c) пересмотр политики в области качества и целей качества;
d) документальное оформление результатов проверки, проведенной руководством, и предпринятых действий, а также своевременный и эффективный обмен информацией о них, включая информирование высшего руководства о соответствующих результатах.
5. Термины и определения
Термины ICH и ISO используются в настоящем документе там, где в них возникает необходимость. В случае, если в контексте настоящей главы слова «требование», «требования» или «необходимо» появляются в определении ISO, то они необязательно отражают нормативные требования. Если не было возможности применить определения ICH или ISO, разработаны формулировки для использования их именно в настоящей главе.
Для целей настоящей главы используются понятия, которые означают следующее:
«возможность процесса» (capability of a process) - способность процесса создавать продукцию, которая будет соответствовать требованиям к этой продукции. Концепция возможности процесса также может определяться при помощи статистических терминов;
«высшее руководство» (senior management) - лицо (лица), которое руководит и осуществляет на высшем уровне контроль за деятельностью предприятия или за его отдельной площадкой, а также имеет полномочия и несет ответственность, чтобы задействовать ресурсы предприятия или его отдельной площадки;
«деятельность, передаваемая для выполнения другой организации (аутсорсинг) » (out sourced activities) - деятельность, осуществляемая подрядчиком по письменному договору с заказчиком;
«инновация» (innovation) - внедрение новых технологий или методологий;
«качество» (quality) - степень, в которой совокупность неотъемлемых свойств продукции, системы или процесса соответствует требованиям;
«контролируемое состояние» (state of control) - условие, при котором комплекс контрольных мероприятий обеспечивает стабильную эффективность процесса и качество продукции;
«корректирующие действие» (corrective action) - действие, направленное на устранение причины выявленного несоответствия или другой нежелательной ситуации. Корректирующее действие осуществляется для предотвращения повторения события;
«обратная связь» (feed-back) - модификация или контроль процесса либо системы исходя из их реально полученных результатов или эффективности. Обратную связь можно технически использовать в стратегии контроля процесса и концептуально - в управлении качеством;
«планирование качества» (quality planning) - часть управления качеством, направленная на установление целей качества и определение необходимых операционных процессов и соответствующих ресурсов для достижения этих целей;
«показатели эффективности» (performance indicators) - измеряемые значения, используемые для количественного выражения целей качества, отражающих эффективность организации, процесса и системы. В некоторых регионах используется термин «метрика эффективности»;
«политика в области качества» (quality policy) - общие цели и направление деятельности организации относительно качества, которые официально формулируются высшим руководством;
«постоянное улучшение» (continual improvement) - постоянная деятельность, направленная на увеличение способности соответствовать требованиям;
«предупреждающее действие» (preventive action) - действие, направленное на устранение причин потенциальных несоответствий или других нежелательных ситуаций. Предупреждающее действие осуществляется для предотвращения события;
«пространство проектных параметров» (design space) - многомерная комбинация и взаимодействие различных исходных переменных (например, свойств материалов), а также параметров процесса, при которых доказано обеспечение качества;
«прямая связь» (feed-forward) - модификация или контроль процесса исходя из их прогнозируемых результатов или эффективности. Прямую связь можно технически использовать в стратегии контроля процесса и концептуально - в управлении качеством;
«реализация продукции» (product realisation) - достижение производства продукции с показателями качества, соответствующими потребностям пациентов, медицинских работников, уполномоченных органов (включая соответствие установленным требованиям), а также внутренних потребителей;
«руководство по качеству» (quality manual) - документ, содержащий систему управления качеством, применяемую в организации;
«средство улучшения» (enabler) - инструмент или процесс, обеспечивающее средства для достижения цели;
«стратегия контроля» (control strategy) - запланированный комплекс мероприятий по контролю, основанный на понимании данной продукции и процесса, и обеспечивающие эффективность процесса и качество продукции. Стратегия контроля может включать в себя контроль параметров и характеристик, относящихся к активным фармацевтическим субстанциям, исходному сырью и компонентам лекарственных препаратов, условиям эксплуатации помещений и оборудования, контролю в процессе производства, спецификациям на готовую продукцию, а также связанные методы и частоту проведения мониторинга и контроля;
«управление знаниями» (knowledge management) - систематический подход к сбору, анализу, накоплению и распространению информации о продукции, производственных процессах и компонентах;
«управление изменениями» (change management) - систематический подход к предложению, оценке, утверждению, внедрению и проверке изменений;
«управление рисками для качества» (quality risk management) - систематический процесс для общей оценки, контроля, передачи информации и обзора рисков для качества лекарственных препаратов на протяжении жизненного цикла продукции;
«фармацевтическая система качества» (ФСК) (pharmaceutical quality system, PQS) - система управления для направления и контроля фармацевтической компании в отношении качества;
«цели качества» (quality objectives) - средства преобразования политики в области качества и стратегий в измеряемую деятельность.
ДОПОЛНЕНИЕ № 3
к Правилам надлежащей
производственной практики
Евразийского экономического союза
Потенциальные возможности улучшения научных
и основанных на анализе рисков надзорных подходов
|
План действий |
Потенциальная возможность |
|
1. Соответствовать требованиям Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила) |
соответствие - обязательно |
|
2.Демонстрировать эффективную фармацевтическую систему качества, включая эффективное использование принципов управления рисками для качества (например, настоящий документ и глава II части III Правил «У правление рисками для качества»). |
увеличение использования подходов, основанных на анализе рисков, при проведении инспекций уполномоченными органами |
|
3. Демонстрировать понимание продукции и процесса, включая эффективное использование принципов управления рисками для качества (например, документ «Управление рисками для качества» настоящих Правил и документ «Фармацевтическая разработка» (ICH Q8). |
содействие научно обоснованной оценке фармацевтического качества обеспечение возможности применения инновационных подходов к процессу валидации внедрение механизмов выдачи разрешения на выпуск в реальном времени |
|
4. Демонстрировать эффективную фармацевтическую систему качества и понимание продукции и процесса, включая эффективное использование принципов управления рисками для качества (например, настоящий документ, документ «Фармацевтическая разработка» (ICH Q8) и глава II части III настоящих Правил) |
увеличение использования подходов, основанных на анализе рисков, при проведении инспекций уполномоченными органами содействие научно-обоснованной оценке фармацевтического качества оптимизация научно обоснованных и основанных на анализе рисков процессов изменений, вносимых после регистрации лекарственных средств, для извлечения максимальной пользы от инноваций и постоянного улучшения обеспечение возможности применения инновационных подходов к процессу валидации внедрение механизмов выдачи разрешения на выпуск в реальном времени |
Настоящий документ отражает потенциальные возможности улучшения подходов, которые могут применяться уполномоченными органами (организациями) государств-членов. Фактический надзорный процесс определяется законодательством соответствующего государства-члена.
ДОПОЛНЕНИЕ № 4
к Правилам надлежащей
производственной практики
Евразийского экономического союза
СХЕМА
модели фармацевтической системы качества
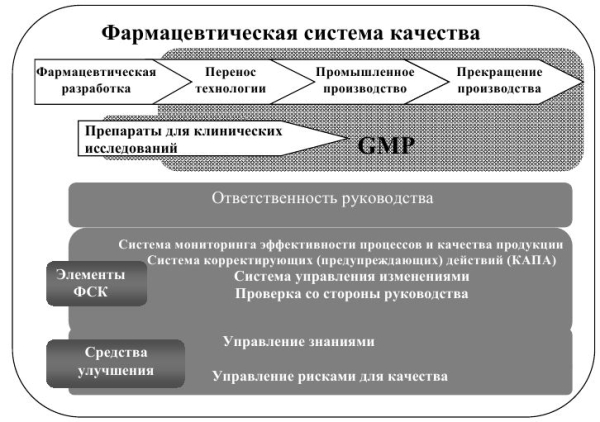
На схеме показаны основные особенности модели фармацевтической системы качества (далее - ФСК). ФСК охватывает весь жизненный цикл продукции, включая фармацевтическую разработку, перенос технологии, промышленное производство и прекращение производства.
ФСК расширяет требования Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила), как показано на схеме. Схема также показывает то, чтотребования Правил распространяются на производство лекарственных препаратов для клинических исследований.
Второй горизонтальный блок иллюстрирует важность ответственности руководства (которая разъясняется в разделе 2 главы III части III Правил) в отношении всех этапов жизненного цикла продукции. В следующем за ним горизонтальном блоке приведен перечень элементов ФСК, которые являются опорными для модели ФСК. Эти элементы следует соответственно и пропорционально применять к каждому этапу жизненного цикла продукции, осознавая возможности выявления сфер для постоянного улучшения.
В наборе горизонтальных блоков в нижней части схемы показаны средства улучшения: управление знаниями и управление рисками для качества, которые применяются на протяжении всех этапов жизненного цикла продукции. Эти средства улучшения помогают при достижении целей ФСК в отношении производства продукции, соответствующей своему предназначению, установления и поддержания контролируемого состояния и содействуют постоянному улучшению.
Глава IV. Международные гармонизированные требования
к сертификации серии
Основные требования
В настоящей главе изложены международные гармонизированные требования к содержанию сертификата серии лекарственного средства.
Каждую серию лекарственного средства, в том числе перемещаемую между странами, в которых соблюдаются требования надлежащего производства, необходимо сопровождать сертификатом серии, выданным производителем лекарственного средства.
Такой сертификат выдается после полного качественного и количественного анализа всех действующих веществ и иных соответствующих ингредиентов для гарантии того, что качество лекарственного средства соответствует всем требованиям регистрационного досье. Сертификат серии должен подтверждать, что серия соответствует спецификациям и произведена в соответствии с регистрационным досье. В сертификате должны быть приведены подробные спецификации на лекарственное средство, ссылки на аналитические методы, полученные результаты аналитических испытаний, а также должно содержаться подтверждение того, что записи по производству, упаковке и контролю качества серии проверены и сделан вывод об их соответствии требованиям настоящих Правил. Сертификат серии должен быть подписан лицом, ответственным за подтверждение того, что серия пригодна к выпуску в реализацию или для поставок (экспорта).
Организация, реализующая лекарственное средство, должна получить и сохранять выданный производителем сертификат. По запросу этот сертификат должен быть легкодоступен сотрудникам уполномоченных органов. Такой сертификат производителя о соответствии серии может иметь особое значение для того, чтобы не проводить повторный контроль, если это разрешено соответствующими нормативными правовыми актами.
При необходимости такой сертификат также может быть выдан для такой продукции, не являющейся готовым лекарственным препаратом, как промежуточная продукция, нерасфасованная или частично упакованная продукция.
Такой сертификат также может быть выдан на активные фармацевтические субстанции и исследуемые лекарственные препараты, используемые в клинических исследованиях. Рекомендуется использовать терминологию, которая приведена в разделе «Определения» настоящей главы.
Сертификат серии лекарственного средства оформляется на бланке и должен содержать следующие сведения:
1. Название продукции
2. Страна-импортер
3. Номер регистрационного удостоверения или номер разрешения на проведение клинических исследований
4. Дозировка (активность)
5. Лекарственная форма
6. Размер и тип упаковки
7. Номер серии
8. Дата производства
9. Дата окончания срока годности
10. Названия, адреса и номера лицензий всех производственных площадок и мест проведения контроля качества
11. Сертификаты соответствия требованиям Правил надлежащей производственной практики Евразийского экономического союза для всех площадок, приведенных в пункте 10
12. Результаты анализов
13. Комментарии
14. Заявление о сертификации
15. Фамилия и должность (звание) лица, выдавшего разрешение на выпуск серии
16. Подпись лица, выдавшего разрешение на выпуск серии
17. Дата подписания
Пояснения и определения
1. Название продукции
Указывается патентованное название, торговое название или присвоенное название в стране-импортере (в зависимости от того, что применимо). В случае лекарственных препаратов для клинических исследований - номер кода, содержащийся в заявке на клинические исследования.
2. Страна-импортер
Указывается наименование государства, осуществляющего импорт серии лекарственного средства.
3. Номер регистрационного удостоверения или номер разрешения на проведение клинических исследований
Указывается номер регистрационного удостоверения на лекарственное средство в стране-импортере. Для лекарственных препаратов для клинических исследований - номер разрешения на проведение клинических исследований или ссылка на исследования (при наличии).
4. Дозировка (активность)
Указываются название и количество в единице лекарственной формы всех активных фармацевтических субстанций (компонентов). В случае лекарственных препаратов для клинических исследований, включая плацебо, способ предоставления такой информации не должен способствовать раскодированию «слепого» исследования.
5. Лекарственная форма
Указывается лекарственная форма (например, таблетки, капсулы, мази).
6. Размер и тип упаковки
Указывается вместимость контейнера и его тип (например, ампулы, флаконы, блистеры и т.п.).
7. Номер серии
Указывается номер серии или номер партии, относящийся к продукции. Уникальная комбинация цифр, букв или символов, идентифицирующая серию, по которой можно проследить историю производства и оптовой торговли серии.
8. Дата производства
Указывается в соответствии с национальными (региональными) требованиями страны-импортера.
9. Дата окончания срока годности
Указывается размещенная на контейнере (этикетке) дата для определения времени, на протяжении которого ожидается, что продукция при ее хранении в требуемых условиях соответствует утвержденным в стране-импортере спецификациям, действующим в течение всего срока годности; после этой даты продукцию применять не следует.
10. Названия, адреса и номера лицензий всех производственных площадок и мест проведения контроля качества
Указываются все производственные площадки, связанные с данным производством, включая упаковку (маркировку) и контроль качества серии с указанием названия, адреса и номера лицензии.
Названия и адреса должны соответствовать указанным в лицензии на производство.
11. Сертификаты соответствия требованиям настоящих Правил для всех производственных площадок, указанных в сертификате лекарственного средства.
Указываются номера сертификатов.
12. Результаты анализов
Указываются утвержденные спецификации, все полученные результаты и ссылки на примененные методы (можно привести ссылку на отдельный подписанный и датированный сертификат анализа, который должен быть приложен).
13. Комментарии
Указывается какая-либо дополнительная информация, которая может быть полезной для импортера и (или) инспектора, подтверждающего соответствие серии (например, специальные условия хранения или транспортирования).
14. Заявление о сертификации
Заявление должно охватывать производство, включая упаковку (маркировку) и контроль качества. Необходимо использовать следующий текст: «Настоящим я подтверждаю, что приведенная выше информация является достоверной и точной. Эта серия продукции произведена (включая упаковку (маркировку) и контроль качества) на вышеуказанной производственной площадке (площадках) в полном соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза и требованиями контроля качества лекарственных средств, а также в соответствии со спецификациями, содержащимися в регистрационном досье страны- импортера или в досье спецификаций на лекарственный препарат в случае лекарственных препаратов для клинических исследований.
Записи по производству, упаковке и анализу проверены, и установлено их соответствие требованиям Правил надлежащей производственной практик Евразийского экономического союза».
15. Фамилия и должность (звание) лица, выдавшего разрешение на выпуск серии
Указываются фамилия и должность (звание) лица, выдавшего разрешение на выпуск серии. Если в пункте 10 приведено больше 1 площадки, также указываются название и адрес площадки.
16. Подпись лица, выдавшего разрешение на выпуск серии Указывается подпись лица, выдавшего разрешение на выпуск серии.
17. Дата подписания
Указывается дата подписания сертификата лицом, выдавшим разрешение на выпуск серии.
Словарь эквивалентных терминов, используемых в сертификате серии лекарственного средства
Настоящий словарь не является исчерпывающим.
Действующие вещества - активные фармацевтические субстанции (АФС) (компоненты) (active substances - active pharmaceutical ingredients (constituents)).
Серия - партия (batch - lot).
Лекарственная форма (dosage form - pharmaceutical form). Производитель (manufacturer - fabricator).
Производство (manufacturing (manufacture) - fabrication).
Лицензия на производство (manufacturing authorisation - establishment license).
Лекарственное средство (medicinal product - pharmaceutical product drug product).
Контроль качества - испытания (quality control - testing).
ПРИЛОЖЕНИЕ № 1
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству стерильных лекарственных средств
Принцип
К производству стерильных лекарственных средств предъявляются особые требования, чтобы свести к минимуму риск контаминации микроорганизмами, частицами и пирогенами. При этом многое зависит от квалификации производственного персонала, его обучения и отношения к работе. Особо важное значение имеет обеспечение качества. При производстве стерильных препаратов необходимо строго придерживаться тщательно разработанных и валидированных способов производства и процедур. Никакая конечная стадия производства или контроль готовой продукции не может рассматриваться как единственное средство обеспечения стерильности или других показателей качества продукции.
Правила надлежащей производственной практики Евразийского экономического союза не устанавливают детальные методы определения чистоты воздуха, поверхностей и пр. по микроорганизмам и частицам. Дополнительную информацию можно получить из других нормативных и технических документов.
Общие требования
1. Стерильную продукцию необходимо производить в чистых зонах, доступ в которые персонала и (или) поступление оборудования, исходного сырья и материалов должны осуществляться через воздушные шлюзы. В чистых помещениях (зонах) должен поддерживаться уровень чистоты, отвечающий соответствующему стандарту чистоты, в них необходимо подавать воздух, который прошел через фильтры соответствующей эффективности.
2. Различные операции по подготовке компонентов, приготовлению продукции и наполнению следует осуществлять в отдельных зонах (помещениях) внутри чистой зоны (помещения). Технологические операции делятся на 2 категории:
а) при которой продукцию подвергают финишной стерилизации (в первичной упаковке);
б) при которой операции на нескольких или всех стадиях выполняют в асептических условиях.
3. Чистые помещения (зоны) для производства стерильной продукции классифицируются в соответствии с требуемыми характеристиками производственной среды. Каждая производственная операция требует определенного уровня чистоты производственной среды в эксплуатируемом состоянии с целью сведения к минимуму риска контаминации частицами или микроорганизмами продукта или обрабатываемого исходного сырья и материалов.
Для обеспечения соответствия требованиям в «эксплуатируемом состоянии» (состояние при котором чистое помещение (зона) и технологическое оборудование функционируют в требуемом режиме с заданным количеством работающего персонала) эти помещения (зоны) должны быть спроектированы так, чтобы обеспечить точно определенный уровень чистоты воздуха в «оснащенном состоянии» (состояние, в котором чистое помещение построено и функционирует, технологическое оборудование полностью укомплектовано, но персонал отсутствует).
Требования к «оснащенному» и «эксплуатируемому» состоянию должны быть установлены для каждого чистого помещения (зоны) или комплекса чистых помещений (зон).
Чистые помещения (зоны) при производстве стерильных лекарственных средств подразделяются на 4 класса:
класс A - локальное помещение (зона) для проведения операций, представляющих высокий риск для качества продукции, например, помещения (зоны) наполнения, укупорки, помещения (зоны), где ампулы и флаконы находятся в открытом состоянии и выполняются соединения частей оборудования в асептических условиях. Как правило, такие условия обеспечиваются ламинарным потоком воздуха на рабочем месте. Системы ламинарного потока воздуха должны обеспечивать равномерную скорость воздуха в диапазоне 0,36 - 0,54 м/с (нормативное значение) в рабочей зоне открытого чистого помещения. Поддержание ламинарности должно быть доказано и провалидировано. В закрытых изоляторах и боксах с перчатками допускается использовать однонаправленный поток воздуха с меньшими скоростями;
класс B - помещение (зона), непосредственно окружающее зону класса A, предназначенную для асептического приготовления и наполнения;
классы C и D - чистые помещения (зоны) для выполнения менее критичных стадий производства стерильной продукции.
Классификация чистых помещений (зон)
4. Чистые помещения и чистые зоны следует классифицировать в соответствии с межгосударственным стандартом ГОСТ ИСО 14644-1 «Чистые помещения и связанные с ними контролируемые среды». Подтверждение класса чистоты следует четко отделять от мониторинга производственной среды при проведении процесса. Максимально допустимая концентрация аэрозольных частиц для каждого класса приведена в таблице 1.
Таблица 1
|
Помещение (зона) |
Максимально допустимое число частиц в 1 куб. м воздуха при размере частиц равном или большем |
|||
|
в оснащенном состоянии |
в эксплуатируемом состоянии |
|||
|
0,5 мкм |
5,0 мкм |
0,5 мкм |
5,0 мкм |
|
|
A |
3 520 |
20 |
3 520 |
20 |
|
B |
3 520 |
29 |
352 000 |
2 900 |
|
C |
352 000 |
2 900 |
3 520 000 |
29 000 |
|
D |
3 520 000 |
29 000 |
не регламентируется |
не регламентируется |
³ 5,0 мкм. Класс B (в оснащенном состоянии) по количеству аэрозольных частиц соответствует классу ИСО 5 по количеству частиц обоих указанных размеров. Класс C (в оснащенном и эксплуатируемом состояниях) по количеству аэрозольных частиц соответствует классу ИСО 7 и ИСО 8 соответственно. Класс D (в оснащенном состоянии) по количеству аэрозольных частиц соответствует классу ИСО 8. Для подтверждения класса в стандарте ГОСТ ИСО 14644-1 установлена методика, в которой регламентированы как минимальное количество точек для отбора проб, так и объем пробы с учетом пределов данного класса для количества частиц наибольшего из указанных размеров, а также метод оценки полученных данных.
6. Для подтверждения класса следует использовать портативные счетчики частиц с короткими трубками для отбора проб из-за относительно высокого уровня осаждения частиц размером ³ 5,0 мкм в дистанционных системах для отбора проб с длинными трубками. В случае систем однонаправленного потока воздуха следует использовать изокинетические насадки для отбора проб.
7. Подтверждение класса в эксплуатируемом состоянии допустимо проводить во время работы, или при моделировании рабочих операций, или при наполнении питательными средами, как того требует моделирование «наихудшего случая». Указания по проведению испытаний для подтверждения постоянного соответствия заданному классу чистоты приведены в стандартах государств - членов Евразийского экономического союза (далее соответственно - государства-члены, Союз) серии ИСО 14644-2.
Мониторинг чистых помещений (зон)
³ 5,0 мкм непосредственно в месте наполнения в процессе наполнения из-за образования частиц или капель из самого продукта.
10. Рекомендуется, чтобы система мониторинга частиц для помещений (зон) класса А применялась для помещений (зон) класса В, хотя частота отбора проб может быть меньшей. Важность системы мониторинга частиц должна быть определена эффективностью разделения между расположенными рядом зонами классов А и В. В зонах класса В следует проводить мониторинг с такой частотой и соответствующим объемом отбираемых проб, чтобы можно было зафиксировать изменения уровня контаминации и любые ухудшения работы системы, а в случае выхода за уровень тревоги можно было бы принять экстренные меры.
11. Системы мониторинга аэрозольных частиц могут состоять из независимых счетчиков частиц, из системы последовательно расположенных точек отбора проб, присоединенных трубопроводом к одному счетчику частиц, или объединять эти два подхода. При выборе систем контроля следует учитывать требования к размерам частиц. При использовании удаленных систем отбора проб следует учитывать длину трубок и радиусы изгибов трубок с учетом возможности оседания частиц в трубках. При выборе системы мониторинга следует также учитывать любой риск, исходящий от материалов, используемых в технологическом процессе (например, наличие живых микроорганизмов или радиоактивных лекарственных препаратов).
12. При использовании автоматизированной системы текущего мониторинга объем проб зависит, как правило, от скорости отбора проб используемой системы. Объем проб при текущем мониторинге может отличаться от объема проб при проведении квалификации чистых помещений и чистых зон.
13. В зонах класса А и В мониторинг концентрации частиц размером ³ 5,0 мкм имеет особое значение, поскольку это является важным инструментом диагностики для раннего выявления несоответствия. Иногда показатели количества частиц размером ³ 5,0 мкм могут быть ошибочными из-за электронного шума, постороннего света, случайного стечения обстоятельств и т. п. Однако если счетчик последовательно и систематически регистрирует малое число частиц, то это указывает на возможность контаминации, что требует расследования. Такие случаи могут заблаговременно указывать на неисправность системы вентиляции и кондиционирования, установки наполнения или свидетельствовать о нарушении правил во время наладки оборудования или его эксплуатации.
14. Допустимое количество частиц для оснащенного состояния, указанное в таблице 1, должно достигаться после короткого периода восстановления 15 - 20 мин (нормативное значение) при отсутствии персонала после завершения работы.
15. Мониторинг зон классов C и D в эксплуатируемом состоянии следует осуществлять в соответствии с принципами управления рисками для качества. Требования к уровню тревоги и уровню действия будут зависеть от характера выполняемых операций, однако должно быть достигнуто рекомендованное значение «периода восстановления».
16. Другие показатели (например, температура и относительная влажность) зависят от продукции и характера выполняемых операций. Эти параметры не должны влиять на установленные нормативы чистоты.
17. Примеры операций, которые следует выполнять в зонах с разными классами чистоты, приведены в таблице 2 (в соответствии с пунктами 28-35 настоящего Приложения).
Таблица 2
|
Класс |
Для продукции, подлежащей финишной стерилизации (примеры операций) (в соответствии с пунктами 28-30 настоящего приложения) |
|
A |
наполнение продукции, которую нельзя подвергать риску контаминации |
|
C |
приготовление растворов, которые нельзя подвергать риску контаминации; наполнение продукции |
|
D |
приготовление растворов и подготовка первичной упаковки, материалов для последующего наполнения |
Таблица 3
|
Класс |
Для приготовления в асептических условиях (примеры операций) (в соответствии с пунктами 31-35 настоящего приложения) |
|
A |
асептическое приготовление и наполнение |
|
C |
приготовление растворов, подлежащих фильтрации |
|
D |
операции с материалами после мойки |
18. При выполнении асептических процессов необходимо часто проводить микробиологический мониторинг с использованием седиментационного и аспирационного методов отбора проб воздуха, отбора проб с поверхностей методом смывов тампоном и с использованием контактных пластин. Методы отбора проб, используемые в эксплуатируемом состоянии, не должны наносить вред защите зоны. Результаты мониторинга следует учитывать при проведении обзора записей производства серии (досье на серию) для выдачи разрешения на выпуск готовой продукции. После выполнения критических операций следует проводить мониторинг поверхностей и персонала. Следует также проводить дополнительный микробиологический мониторинг вне технологического процесса (например, после валидации систем, очистки и дезинфекции).
19. Рекомендуемые пределы при микробиологическом мониторинге чистых помещений (зон) в эксплуатируемом состоянии приведены в таблице 4.
Таблица 4
|
Класс |
Рекомендуемые пределы микробной контаминации* |
|||
|
в воздухе, КОЕ/м3 |
седиментация на чашку диаметром 90 мм, КОЕ за 4 ч ** |
контактные пластины диаметром 55 мм, КОЕ/пластина |
отпечаток перчатки (5 пальцев), КОЕ/перчатка |
|
|
A |
< 1 |
< 1 |
< 1 |
< 1 |
|
B |
10 |
5 |
5 |
5 |
|
C |
100 |
50 |
25 |
- |
|
D |
200 |
100 |
50 |
- |
*Приведены средние значения.
**Отдельные пластины для седиментации могут экспонироваться менее 4 часов.
20. По результатам мониторинга частиц и микроорганизмов должны быть установлены соответствующие пределы: уровень тревоги и уровень действия. В операционных процедурах должны быть описаны корректирующие действия в случае превышения этих пределов.
Изолирующая технология
21. Использование изолирующей технологии сводит к минимуму вмешательство человека в производственных зонах, в результате чего значительно снижается риск микробной контаминации продукции, произведенной в асептических условиях, из производственной среды. На фармацевтическом производстве допускается использование различных типов изоляторов и передаточных устройств. При этом изолятор и его комплектующие должны быть сконструированы таким образом, чтобы в соответствующей зоне обеспечивалось необходимое качество воздуха. Следует учитывать, что изоляторы, изготовленные из разных материалов, в различной степени подвержены повреждению изоляции и разгерметизации. Допускается использование различных передаточных устройств: от конструкций с одинарной или двойной дверью до полностью герметизированных систем, включающих устройства для стерилизации.
22. Передача материалов внутрь и наружу изолятора является одним из основных потенциальных источников контаминации. Как правило, пространство внутри изолятора является ограниченной зоной для проведения операций, представляющих высокие риски для качества продукции. Допускается, что в рабочей зоне всех таких устройств может отсутствовать ламинарный поток воздуха.
23. Требования к чистоте воздуха в среде, окружающей изолятор, зависят от конструкции изолятора и его назначения. Чистоту этой среды следует контролировать, и для асептического производства она должна быть не ниже класса чистоты D.
24. Изоляторы могут быть введены в эксплуатацию только после проведения соответствующей валидации. Валидация должна учитывать все критические факторы изолирующей технологии (например, качество воздуха внутри и снаружи изолятора, порядок дезинфекции изолятора, процессы передачи и целостность изолятора).
25. Необходимо проводить непрерывный мониторинг, включающий в себя частые испытания герметичности изолятора и узлов «перчатки - рукава».
Технология «выдувание - наполнение - герметизация»
26. Устройство для «выдувания-наполнения-герметизации» представляет собой устройство специальной конструкции, где в одном автоматическом комплексе в течение одного непрерывного технологического цикла из термопластичного гранулята формируются упаковки, которые наполняются продуктом и герметизируются. Устройство для «выдувания - наполнения - герметизации», используемое в асептическом производстве и имеющее зону класса A с эффективным потоком воздуха, может быть установлено, по крайней мере, в зоне класса C при условии использования одежды, применяемой в зонах классов A и (или) B. Производственная среда в оснащенном состоянии должна соответствовать установленным нормативам по частицам и микроорганизмам, а в эксплуатируемом состоянии - только по микроорганизмам. Устройство для «выдувания - наполнения - герметизации», используемое в производстве продукции, подлежащей финишной стерилизации, должно устанавливаться по крайней мере в зоне класса D.
27. Учитывая особенности технологии «выдувание - наполнение - герметизация», необходимо обращать особое внимание на:
конструкцию и квалификацию оборудования;
валидацию и воспроизводимость процессов «очистка на месте» и «стерилизация на месте»;
пространство чистого помещения, которое является производственной средой для размещенного там оборудования;
обучение операторов и их одежду;
действия в критической зоне оборудования, включая выполнение подсоединений и сборки в асептических условиях до начала наполнения.
Продукция, подвергаемая финишной стерилизации
28. Подготовка компонентов первичной упаковки и других материалов и производство большинства видов продукции должны проводиться в производственной среде по крайней мере класса D, чтобы обеспечить достаточно низкий уровень рисков контаминации частицами и микроорганизмами, подходящий для фильтрации и стерилизации.
Если микробная контаминация представляет высокие или особенные риски для продукции (например, когда продукция является хорошей питательной средой для роста микроорганизмов, либо ее стерилизации предшествует длительный период времени, либо технологический процесс ведется по большей части в открытых емкостях), приготовление следует осуществлять в производственной среде класса С.
29. Наполнение продуктами, подлежащими финишной стерилизации, должно проводиться в производственной среде, по крайней мере, класса C.
30. При повышенном риске контаминации продукта от производственной среды (например, если операции наполнения проходят медленно или упаковки имеют широкое горло, или их необходимо держать открытыми более нескольких секунд до герметизации) наполнение должно проводиться в зоне класса A с производственной средой, по крайней мере, класса C. Приготовление и фасовку мазей, кремов, суспензий и эмульсий перед финишной стерилизацией необходимо, как правило, осуществлять в производственной среде класса С.
Асептическое производство
31. Операции с компонентами первичной упаковки и другими материалами после мойки должны проводиться в производственной среде, по крайней мере, класса D. Обработку стерильного исходного сырья и компонентов, если в дальнейшем не предусмотрена стерилизация или стерилизующая фильтрация, следует осуществлять в рабочей зоне класса А с производственной средой класса В.
32. Приготовление растворов, которые в ходе технологического процесса подлежат стерилизующей фильтрации, должно проводиться в производственной среде класса C. Если стерилизующая фильтрация не проводится, то подготовку материалов и производство продукции необходимо осуществлять в рабочей зоне класса А с производственной средой класса В.
33. Проводить обработку и наполнение продукции, приготовленной в асептических условиях, следует в рабочей зоне класса А с производственной средой класса В.
34. Передачу (транспортировку) не окончательно укупоренных первичных упаковок с продукцией (например, лиофилизированной) до завершения процесса укупорки необходимо осуществлять в зоне класса А, находящейся в производственной среде класса B, или в герметичных передаточных контейнерах в производственной среде класса B.
35. Приготовление и наполнение стерильных мазей, кремов, суспензий и эмульсий необходимо осуществлять в зоне класса А, находящейся в производственной среде класса B, если продукция находится в открытых емкостях и в дальнейшем не подвергается стерилизующей фильтрации.
Персонал
36. В чистых помещениях (зонах) допускается нахождение только минимально необходимого количества персонала; что особенно важно для асептического производства. Проверки и контрольные операции следует проводить, по возможности, находясь за пределами чистых помещений (зон).
37. Весь персонал (в том числе персонал, занятый очисткой и техническим обслуживанием), работающий в чистых помещениях (зонах), должен проходить регулярное обучение по вопросам надлежащего производства стерильной продукции, включая вопросы гигиены и основы микробиологии. Если необходимо, чтобы посторонние сотрудники, не прошедшие такого обучения (например, работающие по контракту строители или наладчики оборудования), находились в чистом помещении, то они должны быть подробно проинструктированы, и за ними должно быть установлено строгое наблюдение.
38. Вход в помещения (зоны) стерильного производства персонала, работающего с сырьем из тканей животных или культурами микроорганизмов, которые не используются в текущем технологическом процессе, не допускается, если персоналом не соблюдены четко установленные процедуры в отношении входа в помещения (зоны) стерильного производства.
39. Необходимо выполнять требования к личной гигиене и чистоте. Персонал, занятый в производстве стерильных лекарственных средств, должен быть проинструктирован о том, что он обязан сообщать о любых обстоятельствах, которые могут быть причиной распространения аномального количества или видов контаминантов. При возникновении таких обстоятельств необходимы периодические медицинские осмотры сотрудников. Действия, которые необходимо предпринять в отношении персонала, который может стать источником микробной контаминации, должны определяться специально назначенным компетентным лицом.
40. В чистых зонах запрещается носить наручные часы и ювелирные украшения, а также использовать косметику.
41. Переодеваться и мыться необходимо в соответствии с инструкциями, принятыми в форме письменного документа и разработанными так, чтобы свести к минимуму риск контаминации одежды для работы в чистых зонах или внесения контаминантов в чистые зоны.
42. Одежда и ее качество должны соответствовать технологическому процессу и классу рабочей зоны. Ее нужно носить так, чтобы обеспечить защиту продукции от контаминации.
43. Для работы в чистых помещениях (зонах) класса D волосы, борода и усы (при наличии) персонала должны быть закрыты, следует носить обычный защитный костюм и соответствующую обувь или бахилы. Должны быть приняты соответствующие меры для предотвращения любой контаминации чистой зоны извне.
Для работы в чистых помещениях (зонах) класса С волосы, борода и усы (при наличии) должны быть закрыты, следует носить комбинезон или брючный костюм, плотно облегающий запястья и имеющий высокий воротник, а также соответствующую обувь или бахилы. От одежды и обуви практически не должны отделяться волокна или частицы.
Для работы в чистых помещениях (зонах) классов А или В головной убор должен полностью закрывать волосы, а также бороду и усы (при наличии) и должен быть вставлен в воротник костюма, на лице следует носить маску для предотвращения распространения капелек, а также следует носить соответствующим образом простерилизованные и неопудренные резиновые или пластиковые перчатки и простерилизованную или продезинфицированную обувь. Нижние края штанин должны быть заправлены внутрь обуви, а рукава одежды - в перчатки. Защитная одежда практически не должна выделять волокон или частиц и должна задерживать частицы, отделяющиеся от тела.
44. Уличную одежду запрещается вносить в комнаты для переодевания, которые ведут в помещения классов В и С. Каждый сотрудник в зоне классов А и В должен быть обеспечен чистой стерильной (простерилизованной или прошедшей соответствующую санитарную обработку) защитной одеждой на каждую рабочую смену. Перчатки во время работы необходимо регулярно дезинфицировать. Маски и перчатки необходимо менять, по крайней мере, каждую смену.
45. Одежду для чистых помещений необходимо очищать и обрабатывать таким образом, чтобы она впоследствии не становилась причиной контаминации. Эти операции следует выполнять в соответствии с письменными инструкциями. Для подготовки такой одежды желательно иметь отдельные прачечные. Неправильная обработка одежды повреждает волокна ткани, что увеличивает риск отделения частиц.
Помещения
46. В чистых зонах все открытые поверхности должны быть гладкими, непроницаемыми и неповрежденными, чтобы свести к минимуму образование и накопление частиц или микроорганизмов, а также должны позволять многократно применять моющие и, при необходимости, дезинфицирующие средства.
47. Для уменьшения накопления пыли и облегчения уборки в помещениях не должно быть не поддающихся очистке углублений и должно быть как можно меньше выступающих краев, полок, шкафов и оборудования. Двери должны быть сконструированы без углублений, недоступных для очистки; раздвижные двери использовать нежелательно.
48. Подвесные потолки должны быть герметичными с целью предотвращения попадания контаминантов из пространства над ними.
49. Монтаж трубопроводов, воздуховодов и другого оборудования следует выполнять так, чтобы не было углублений и незакрытых отверстий, а также отсутствовали поверхности, не доступные для очистки.
50. Запрещается устанавливать раковины и сливы в зонах классов A или B, используемых для асептического производства. В других зонах следует предусматривать разрыв струи между оборудованием и канализационной трубой (воронкой). Стоки в полу в чистых помещениях с более низким классом чистоты должны быть обеспечены сифонами или гидрозатворами для предотвращения обратного потока.
51. Помещения для переодевания должны быть сконструированы как воздушные шлюзы и должны использоваться для обеспечения физического разделения разных этапов смены одежды и сводить к минимуму контаминацию защитной одежды микроорганизмами и частицами. Они должны эффективно обдуваться отфильтрованным воздухом. Зона перед выходом из помещения для переодевания в оснащенном состоянии должна иметь тот же класс чистоты, что и помещение (зона), в которую она ведет. В некоторых случаях для входа в чистые помещения (зоны) и выхода из них целесообразно иметь отдельные помещения для переодевания. Как правило, устройства для мытья рук должны быть расположены внутри комнат для переодевания недалеко от входа в них.
52. Обе двери воздушного шлюза не могут быть открыты одновременно. Для предотвращения одновременного открывания более одной двери должна работать блокировочная система или система визуального и (или) звукового предупреждения.
53. Подача отфильтрованного воздуха должна поддерживать положительный перепад давления относительно производственных зон с более низким классом при всех рабочих условиях, а воздушный поток должен эффективно обтекать зону. Смежные помещения с разными классами чистоты должны иметь разницу в давлении 10 - 15 Па (нормативное значение). Особое внимание следует уделять защите помещения (зоны) наибольших рисков для качества продукции, то есть производственной среде, непосредственному влиянию которой подвергается продукция или очищенные компоненты, контактирующие с продукцией. Допускаются разные варианты в отношении подачи воздуха и перепада давлений, которые могут потребоваться из-за присутствия некоторых материалов (например, патогенных, высокотоксичных, радиоактивных материалов или живых вирусов, или бактериальных материалов, или препаратов из них). Для некоторых операций может быть необходима деконтаминация помещений и оборудования и обработка воздуха, удаляемого из чистого помещения (зоны).
54. Следует наглядно продемонстрировать, что направление воздушных потоков не представляет рисков для контаминации продукта, например, следует удостовериться, что в помещение (зону), представляющее наибольшие риски для качества продукта, с воздушным потоком не поступают частицы, источниками выделения которых являются обслуживающий персонал, выполняемая операция или оборудование.
55. Следует предусмотреть систему аварийного оповещения об отказе системы вентиляции. Если разница в давлении между двумя помещениями является критичной, между ними необходимо установить датчики перепада давления. Значения перепада давления необходимо регулярно записывать или оформлять документально иным способом.
Оборудование
56. Не допускается, чтобы через перегородку, отделяющую помещение (зону) класса А или В от производственной зоны с более низкой чистотой воздуха, проходила лента конвейера, за исключением случаев, когда сама лента подвергается непрерывной стерилизации (например, в стерилизационном туннеле).
57. Насколько это возможно практически, оборудование, фитинги (места соединения) и зоны обслуживания должны быть спроектированы и установлены таким образом, чтобы работы с оборудованием, его техническое обслуживание и ремонт можно было проводить снаружи чистого помещения (зоны). Если необходима стерилизация, то она должна быть проведена после максимально полной сборки оборудования.
58. Если техническое обслуживание оборудования было проведено внутри чистого помещения (зоны) и необходимые нормы чистоты и (или) асептики были нарушены во время этой работы, то помещение (зона) должно быть очищено, продезинфицировано и (или) простерилизовано (в зависимости от того, что подходит) до возобновления процесса.
59. Установки для подготовки воды и системы ее распределения следует проектировать, конструировать и эксплуатировать так, чтобы обеспечить надежное обеспечение водой соответствующего качества. Их нельзя эксплуатировать сверх проектной мощности. Воду для инъекций необходимо производить, хранить и распределять таким образом, чтобы предотвратить рост микроорганизмов, например, за счет ее постоянной циркуляции при температуре выше 70 0 С.
60. Такое оборудование, как стерилизаторы, системы обработки и фильтрации воздуха, воздушные и газовые фильтры, системы обработки, получения, хранения и распределения воды, должно подлежать валидации и плановому техническому обслуживанию. На его повторное введение в эксплуатацию должно быть выдано разрешение.
Санитария
61. Санитарная обработка чистых помещений (зон) имеет особо важное значение. Помещения (зоны) необходимо тщательно очищать в соответствии с инструкцией, принятой в форме письменного документа. В случае проведения дезинфекции следует применять несколько типов дезинфицирующих средств. Для выявления развития резистентных штаммов микроорганизмов следует проводить регулярный контроль.
62. Моющие и дезинфицирующие средства необходимо контролировать в отношении микробиологической чистоты. Их растворы следует держать в предварительно очищенных контейнерах (таре) и хранить лишь на протяжении установленных сроков, за исключением тех растворов, которые простерилизованы. Моющие и дезинфицирующие средства, используемые в помещениях (зонах) классов А и B, перед использованием должны быть стерильными.
63. Для снижения микробной контаминации в недоступных местах может быть полезна фумигация чистых помещений (зон).
Технологический процесс
64. На всех стадиях производства, в том числе на стадиях, предшествующих стерилизации, необходимо принимать меры, сводящие к минимуму контаминацию.
65. Не допускается производство лекарственных средств микробиологического происхождения или их фасовка (наполнение контейнеров) в помещениях, используемых для производства других лекарственных средств. Вакцины, содержащие убитые микроорганизмы или бактериальные экстракты, после инактивации могут быть расфасованы в тех же помещениях (зонах), что и другие стерильные лекарственные средства.
66. Валидация процессов, проводимых в асептических условиях, должна включать моделирование процесса с использованием питательной среды (наполнение питательными средами). Питательную среду следует выбирать с учетом лекарственной формы лекарственного препарата, а также селективности, прозрачности, концентрации и пригодности питательной среды для стерилизации.
67. Моделирование процесса должно наиболее точно имитировать серийный процесс асептического производства и включать в себя его последовательные критические стадии. Также следует учитывать различные вмешательства, которые могут возникнуть во время обычного производственного процесса, а также ситуации «наихудшего случая».
68. Моделирование процесса при первоначальной валидации должно включать 3 последовательных удовлетворительных испытания для каждой смены операторов. В дальнейшем их следует повторять через установленные промежутки времени, а также после любого существенного изменения в системе вентиляции и кондиционирования воздуха, оборудовании, процессе или количестве смен. Как правило, моделирующие процесс испытания следует повторять 2 раза в год для каждой смены операторов и каждого процесса.
69. Количество контейнеров (первичных упаковок), предназначенных для фасовки питательных сред, должно быть достаточным, чтобы обеспечить достоверную оценку. В случае небольших серий количество контейнеров для фасовки питательных сред должно как минимум соответствовать размеру серии продукции. Необходимо стремиться к отсутствию роста микроорганизмов, при этом следует применять такие нормы:
a) в случае наполнения менее 5 000 единиц продукции, не должно быть ни одной контаминированной единицы;
b) в случае наполнения от 5 000 до 10 000 единиц продукции:
наличие 1 контаминированной единицы является основанием для расследования причин и повторной фасовки питательных сред; наличие 2 контаминированных единиц является основанием для расследования причин и повторной валидации;
в) в случае наполнения свыше 10 000 единиц:
наличие 1 контаминированной единицы является основанием для расследования причин;
наличие 2 контаминированных единиц является основанием для расследования причин и повторной валидации.
70. При любом количестве первичных упаковок с питательной средой периодические случаи обнаружения микробной контаминации могут указывать на наличие небольших уровней контаминантов, что должно быть расследовано. При обнаружении значительной микробной контаминации следует рассмотреть возможное влияние на стерильность серий, выпущенных после проведения последних успешных испытаний с наполнением питательными средами.
71. Необходимо обеспечить условия, при которых любая валидация не создает риска для технологических процессов.
72. Источники водоснабжения, оборудование для подготовки воды и приготовленная вода подлежат регулярному мониторингу на наличие химических и биологических контаминантов и, при необходимости, на наличие эндотоксинов. Результаты мониторинга и любых предпринятых действий следует оформлять документально.
73. В чистых помещениях (зонах), особенно в ходе процесса асептического производства, деятельность персонала должна быть минимальной, а его передвижение должно быть методическим и контролируемым во избежание избыточного выделения частиц и микроорганизмов, обусловленного повышенной двигательной активностью. Температура и влажность производственной среды должны быть не очень высокими, чтобы не создавать дискомфорта с учетом свойств используемой одежды.
74. Микробная контаминация исходного сырья и материалов должна быть минимальной. Спецификации на них должны включать в себя требования к микробиологической чистоте.
75. В чистых помещениях (зонах) необходимо сводить к минимуму наличие контейнеров и материалов, от которых возможно отделение волокон.
76. Следует принимать меры по предотвращению контаминации готовой продукции частицами.
77. По окончании процесса очистки компонентов, контейнеров и оборудования с ними следует обходиться так, чтобы не происходила их повторная контаминация.
78. Интервалы времени между мойкой, сушкой и стерилизацией компонентов, контейнеров и оборудования, а также между их стерилизацией и последующим использованием должны быть минимальными и иметь ограничение по времени, соответствующее условиям хранения.
79. Время между началом приготовления раствора и его стерилизацией или стерилизующей фильтрацией должно быть минимальным. Для каждого вида продукции следует установить максимально допустимое время с учетом его состава и установленного порядка хранения.
80. Перед стерилизацией необходимо контролировать уровень микробной контаминации. Должны быть установлены рабочие границы контаминации непосредственно перед стерилизацией, которые соотносятся с эффективностью используемого метода. Уровень микробной контаминации следует количественно определять для каждой серии продукции, наполненной в асептических условиях, и для каждой серии продукции, подвергаемой финишной стерилизации. Если для лекарственных препаратов, подвергаемых финишной стерилизации, установлены более жесткие параметры стерилизации, уровень микробной контаминации можно контролировать только через соответствующие интервалы времени согласно графику. В случае систем выпуска по параметрам определение микробной контаминации следует проводить для каждой серии и рассматривать как испытание в процессе производства. При необходимости следует контролировать уровень эндотоксинов. Все растворы, особенно инфузионные жидкости большого объема, необходимо подвергать стерилизующей фильтрации по возможности непосредственно перед наполнением.
81. Компоненты, контейнеры, оборудование и любые другие предметы, необходимые в чистом помещении (зоне), при работе в асептических условиях, должны быть простерилизованы и переданы туда через вмонтированный в стену проходной стерилизатор с двусторонним доступом или иным способом, предотвращающим контаминацию. Негорючие газы должны проходить через фильтры, задерживающие микроорганизмы.
82. Эффективность любого нового процесса должна быть подтверждена при валидации, которую необходимо регулярно повторять в соответствии с планом, учитывающим график эксплуатации, а также при любом значительном изменении в процессе или оборудовании.
Стерилизация
83. Все процессы стерилизации должны пройти валидацию. Особое внимание необходимо, если применяемый метод стерилизации не описан в действующем издании соответствующей фармакопеи или используется для продукта, не являющегося простым водным или масляным раствором. Предпочтительным является метод термической стерилизации. В любом случае метод стерилизации должен соответствовать лицензии на производство и регистрационному досье.
84. Перед выбором любого процесса стерилизации необходимо продемонстрировать с помощью физических измерений и, если возможно, биологических индикаторов, что он подходит для данной продукции и эффективен для достижения необходимых условий стерилизации во всех частях каждого типа загрузки. Валидацию процесса необходимо повторять через установленные графиком промежутки, но не реже 1 раза в год, а также всегда в случае внесения существенных изменений в оборудование. Необходимо хранить записи с результатами.
85. Для эффективной стерилизации весь материал в целом должен быть подвергнут необходимой обработке, а процесс организован таким образом, чтобы гарантировать, что должная эффективность будет достигнута.
86. Для всех процессов стерилизации должны быть разработаны и должны пройти валидацию способы загрузки.
87. Применение биологических индикаторов следует рассматривать только как дополнительный метод контроля стерилизации. Биологические индикаторы необходимо хранить и использовать в соответствии с инструкциями производителя, а их качество контролировать методами позитивного контроля. В случае использования биологических индикаторов необходимо принять строгие меры, предотвращающие микробную контаминацию от самих индикаторов.
88. Следует четко определить меры, обеспечивающие разделение продукции, прошедшей и не прошедшей стерилизацию. На каждой корзине, лотке или другой емкости для продукции или компонентов должна быть четкая этикетка с наименованием материала, номером серии и указанием, прошел этот материал стерилизацию или нет. При необходимости могут быть использованы такие индикаторы, как автоклавная лента, для указания того, прошла ли серия (или часть серии) процесс стерилизации, однако они не дают достоверного подтверждения того, серия действительно стерильна.
89. Для каждого цикла стерилизации необходимо составлять записи. Они должны быть утверждены, что является частью процедуры выдачи разрешения на выпуск серии.
Термическая стерилизация
90. Каждый цикл термической стерилизации должен быть записан в виде диаграммы в координатах времени и температуры в достаточно большом масштабе или должен быть зарегистрирован с помощью другого соответствующего оборудования, имеющего необходимую правильность и точность. Место расположения температурных датчиков, используемых для контроля и (или) записи, должно быть определено во время валидации и в случае необходимости также проверено с помощью другого независимого температурного датчика, расположенного в том же месте.
91. Допускается использовать химические и биологические индикаторы, но они не должны заменять проведение физических измерений.
92. Должно быть предусмотрено достаточное время, чтобы весь объем загрузки достиг необходимой температуры до того, как будет начат отсчет времени стерилизации. Этот период должен быть определен для каждого типа стерилизуемой загрузки.
93. После завершения высокотемпературной фазы цикла термической стерилизации должны быть приняты меры предосторожности, предотвращающие контаминацию простерилизованной загрузки во время охлаждения. Любая охлаждающая жидкость или газ, контактирующие с продукцией, должны быть простерилизованы, кроме случаев, когда возможность использования негерметичных упаковок исключена и приведены соответствующие доказательства.
Стерилизация паром
94. При стерилизации паром следует контролировать температуру и давление. Как правило, средства управления должны быть независимы от средств контроля и записывающих устройств. Если для этой цели используются автоматические системы управления и контроля, они должны пройти валидацию, чтобы гарантировать их соответствие требованиям к критическому процессу. Нарушения в ходе процесса должны регистрироваться системой и находиться под надзором оператора. В ходе процесса стерилизации показания независимого датчика температуры следует постоянно сверять с данными диаграммы записывающего устройства. Для стерилизаторов, имеющих сток в дне камеры, может возникнуть необходимость регистрации температуры в этой точке в течение всего цикла стерилизации. Если в цикл стерилизации входит этап вакуумирования, то проверки камеры на герметичность следует проводить с установленной частотой.
95. Стерилизуемые предметы, не находящиеся в герметичных упаковках, должны быть завернуты в материал, пропускающий воздух и пар, но предотвращающий повторную контаминацию этих предметов после стерилизации. Необходимо обеспечить контакт всех частей загрузки со стерилизующим агентом при заданных температуре и времени.
96. Необходимо обеспечить, чтобы для стерилизации применялся пар надлежащего качества, не содержащий такого количества примесей, которое могло бы вызывать контаминацию продукции или оборудования.
Сухожаровая стерилизация
97. При сухожаровой стерилизации должны быть предусмотрены циркуляция воздуха внутри камеры и поддержание избыточного давления для предотвращения попадания внутрь нее нестерильного воздуха. Любой поступающий внутрь воздух должен проходить через фильтры высокой эффективности (НЕРА-фильтр). Если стерилизация предусматривает устранение пирогенов, то как часть валидации должны быть проведены испытания с преднамеренным использованием эндотоксинов.
Радиационная стерилизация
98. Радиационную стерилизацию используют главным образом для стерилизации термочувствительных материалов и продукции. Многие лекарственные средства и некоторые упаковочные материалы чувствительны к ионизирующему излучению, следовательно, этот метод допустим только тогда, когда было экспериментально подтверждено отсутствие вредного влияния на продукцию. Как правило, облучение ультрафиолетовым излучением не является приемлемым методом стерилизации.
99. Во время процесса радиационной стерилизации должно проводиться измерение поглощенной дозы ионизирующего излучения. Для этого следует использовать дозиметры, показания которых не зависят от используемой мощности дозы излучения, но которые обеспечивают количественную регистрацию дозы излучения, поглощенную самой продукцией. Дозиметры должны быть размещены среди загрузки в достаточном количестве и на достаточно близком расстоянии друг от друга, чтобы гарантировать наличие дозиметров во всех местах, подвергаемых облучению. Пластмассовые дозиметры следует применять лишь в пределах срока действия их калибровки. Показания дозиметров необходимо снимать в течение короткого отрезка времени после облучения.
100. В качестве средства дополнительного контроля могут использоваться биологические индикаторы.
101. Процедуры валидации должны гарантировать, что влияние разной плотности укладки стерилизуемой продукции учтено.
102. Процедуры обращения с материалами должны предотвращать перепутывание между облученными и необлученными материалами. На каждую упаковку должны быть нанесены чувствительные к излучению цветовые индикаторы, чтобы различать упаковки, прошедшие и не прошедшие облучение.
103. Суммарная поглощенная доза излучения должна быть набрана в течение времени, отведенного на процесс стерилизации.
Стерилизация оксидом этилена
104. Метод стерилизации оксидом этилена может быть использован только тогда, когда невозможно использование другого способа. Во время валидации процесса должно быть доказано, что отсутствует повреждающее влияние на продукцию, а предусмотренные для дегазации условия и время таковы, что количество остаточного газа и продуктов реакции будет находиться в допустимых пределах, установленных для данного вида продукции или материала.
105. Существенное значение имеет непосредственный контакт между газом и микроорганизмами. Следует принять меры предосторожности от включения микроорганизмов в материал (например, в кристаллы или высушенный белок). Вид и количество упаковочных материалов могут существенно повлиять на процесс.
106. Перед обработкой газом должно быть обеспечено соответствие влажности и температуры материалов требованиям процесса. Требуемое для этого время должно быть, по возможности, минимальным.
107. Каждый цикл стерилизации должен контролироваться с помощью соответствующих биологических индикаторов, необходимое количество которых должно быть равномерно распределено по всей загрузке. Полученная при этом информация должна составлять часть записей, относящихся к серии (досье на серию) готовой продукции.
108. Для каждого цикла стерилизации должны быть оформлены записи с указанием времени полного завершения цикла, давления, температуры и влажности в камере во время процесса, а также концентрации и общего количества использованного газа. Давление и температуру следует регистрировать на протяжении всего цикла на диаграмме. Эти записи должны составлять часть записей, относящихся к серии (досье на серию) готовой продукции.
109. Загрузку после стерилизации следует хранить под контролем в условиях вентиляции, чтобы обеспечить снижение содержания остаточного газа и продуктов реакции до установленного предела. Этот процесс должен пройти валидацию.
Фильтрация лекарственных средств, которые не могут быть
простерилизованы в окончательной упаковке
110. Проведение стерилизующей фильтрации не является достаточным условием стерилизации, если возможно проведение стерилизации продукции в окончательной упаковке. Предпочтительным является метод стерилизации паром. Если продукция не может быть простерилизована в окончательной упаковке, то растворы или жидкости могут быть профильтрованы через стерильный фильтр с номинальным размером пор 0,22 мкм (или менее) или через фильтр с аналогичной способностью задерживать микроорганизмы в предварительно простерилизованные контейнеры (упаковки). Такие фильтры могут удалять большинство бактерий и плесневых грибов, но не все вирусы или микоплазмы. Поэтому должна быть рассмотрена возможность дополнения процесса фильтрации термической обработкой определенной степени.
111. В связи с тем, что при стерилизующей фильтрации по сравнению с другими процессами стерилизации существует потенциальный дополнительный риск, непосредственно перед фасовкой рекомендуется повторная фильтрация через дополнительный стерилизующий фильтр, задерживающий микроорганизмы. Последнюю стерилизующую фильтрацию необходимо осуществлять как можно ближе к месту фасовки.
112. Следует использовать фильтры с минимальным отделением волокон.
113. Перед использованием стерилизующего фильтра и сразу после его использования следует проверять его целостность таким методом, как «точка пузырька», методом диффузионного потока или испытанием под давлением. При валидации следует определять время, необходимое для фильтрации раствора заданного объема, и перепад давлений на фильтре, любые существенные отклонения от этих параметров во время текущего производства следует регистрировать и исследовать. Результаты проверок должны быть включены в состав записей, относящихся к серии (досье на серию) продукции. Сразу после использования следует подтверждать целостность критических газовых и воздушных фильтров. Целостность других фильтров необходимо подтверждать через соответствующие интервалы времени.
114. Не допускается использовать один и тот же фильтр в течение более одного рабочего дня, за исключением случаев, когда возможность более длительного его использования подтверждена валидацией.
115. Фильтр не должен оказывать влияние на продукцию, задерживая ее ингредиенты или выделяя в нее какие-либо вещества.
Окончание процесса производства стерильной продукции
116. Частично укупоренные флаконы после завершения лиофильного высушивания должны находиться в зоне класса A до их полного укупоривания пробкой.
117. Контейнеры (первичные упаковки) должны быть укупорены соответствующими способами, которые прошли валидацию. При использовании метода запайки, например, стеклянных или пластмассовых ампул, вся продукция подлежит 100%-ному контролю на целостность. В других случаях контроль целостности продукции должен проводиться установленными методами.
118. Система укупоривания флаконов, наполненных в асептических условиях, не является полностью целостной до тех пор, пока на укупоренном пробкой флаконе не будет обжат (закатан) алюминиевый колпачок (крышка). В связи с этим обжим колпачка после укупоривания пробкой следует выполнять как можно раньше.
119. Поскольку при обжиме колпачков может выделяться большое количество механических частиц, оборудование для обжима следует размещать отдельно и снабжать его системой вытяжки воздуха.
120. Обжим колпачков на флаконах может проводиться как часть асептического процесса с использованием простерилизованных колпачков или в условиях чистого помещения вне асептической зоны. В последнем случае флаконы должны быть защищены зоной класса А, пока не покинут асептическую зону, и в дальнейшем укупоренные пробками флаконы должны быть защищены путем подачи чистого воздуха класса А, пока на них не будут обжаты колпачки.
121. Флаконы без пробки или со смещенной пробкой следует удалять до обжима колпачков. Если при обжиме колпачков необходимо вмешательство человека, следует использовать соответствующую технологию для исключения прямого контакта с флаконами и минимизации микробной контаминации.
122. Эффективным средством защиты могут быть барьеры или изоляторы, ограничивающие доступ в рабочую зону, обеспечивающие требуемые условия и сводящие к минимуму прямой доступ человека к операции обжима.
123. Первичные упаковки, герметизированные под вакуумом (вакуумные упаковки), должны проверяться на сохранение вакуума после заранее определенного промежутка времени.
124. Первичные упаковки с продукцией для парентерального введения необходимо проверять индивидуально (поштучно) на наличие посторонних включений или других несоответствий по качеству. Визуальный контроль следует проводить при установленных уровнях освещенности и фоне рабочего поля. Следует регулярно проверять зрение операторов, выполняющих визуальный контроль (если операторы используют очки, то проверка зрения проводится в очках). В ходе визуального контроля продукции рекомендуется достаточно часто организовывать перерывы в работе операторов. При использовании других методов контроля процесс контроля необходимо валидировать, состояние оборудования следует периодически проверять. Результаты визуального контроля должны быть оформлены документально.
Контроль качества
125. Испытание готовых продуктов на стерильность необходимо рассматривать только как завершающий этап в серии контрольных мероприятий, гарантирующих стерильность. Методика испытания на стерильность должна быть валидирована для каждого продукта.
126. В случаях если получено разрешение на выпуск стерильной продукции по параметрам, предусмотренным системой выпуска по параметрам согласно приложению № 17 к Правилам надлежащей производственной практики Евразийского экономического союза, следует уделить особое внимание валидации и контролю всего технологического процесса.
127. Выборка образцов продукции, которые были отобраны для проведения испытания на стерильность, должна быть репрезентативной для всей серии и обязательно должна включать образцы, отобранные из тех частей серии, для которых предполагается наибольший риск контаминации, например:
a) для продуктов, наполненных в асептических условиях, образцы должны включать контейнеры (первичные упаковки), в которые происходило наполнение в начале и в конце производства серии, а также после любого значительного вмешательства;
b) для продуктов, прошедших термическую стерилизацию в окончательной упаковке, должно быть уделено внимание отбору проб из потенциально самых холодных частей загрузки.
ПРИЛОЖЕНИЕ № 2
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству биологических (в том числе иммунобиологических)
активных фармацевтических субстанций и лекарственных
препаратов для медицинского применения
Область применения
Технология производства биологических (в том числе иммунобиологических) активных фармацевтических субстанций и лекарственных препаратов для медицинского применения (далее - биологические активные фармацевтические субстанции и лекарственные препараты) является критическим фактором, определяющим соответствующий регуляторный контроль. Поэтому активные фармацевтические субстанции и лекарственные препараты могут быть определены как биологические в значительной мере исходя из технологии их производства. Настоящее приложение служит руководством для всего спектра биологических активных фармацевтических субстанций и биологических лекарственных препаратов.
Настоящее приложение разделено на две основные части:
а) часть A содержит дополнительные правила производства биологических активных фармацевтических субстанций и лекарственных препаратов, начиная с контроля посевных культур и банков клеток и заканчивая заключительными операциями и проведением испытаний;
b) часть B содержит дополнительное руководство для отдельных типов биологических активных фармацевтических субстанций и лекарственных препаратов.
Настоящее приложение наряду с некоторыми другими приложениями содержит указания, дополняющие части I и II Правил надлежащей производственной практики Евразийского экономического союза. Область применения настоящего приложения содержит следующие аспекты:
a) стадия производства: до того момента, пока биологические активные фармацевтические субстанции не станут стерильными, основным документом для них является часть II Правил надлежащей производственной практики Евразийского экономического союза. Правила для последующих стадий производства биологических лекарственных препаратов содержатся в части I Правил надлежащей производственной практики Евразийского экономического союза;
b) вид продукции: настоящее приложение является руководящим документом для всего спектра биологических лекарственных препаратов.
Указанные аспекты представлены в таблице, которая является только иллюстративной и не предназначена для точного описания области применения настоящего приложения. Так же, как и в соответствующей таблице в части II Правил надлежащей производственной практики Евразийского экономического союза, уровень требований усиливается по мере перехода от ранних к поздним стадиям производства биологических активных фармацевтических субстанций, однако принципы Правил надлежащей производственной практики Евразийского экономического союза должны соблюдаться всегда. Включение некоторых ранних стадий производства в область применения настоящего приложения не подразумевает, что эти стадии будут подлежать регулярным проверкам уполномоченными органами (организациями) государств - членов Евразийского экономического союза.
Антибиотики не являются биологическими лекарственными препаратами, однако требования настоящего приложения могут быть использованы на биологических стадиях их производства. Требования к производству лекарственных препаратов, получаемых из фракционированной донорской крови или плазмы, установлены в приложении № 14 к Правилам надлежащей производственной практики Евразийского экономического союза, а для нетрансгенных растительных лекарственных препаратов - в приложении № 7 к Правилам надлежащей производственной практики Евразийского экономического союза.
В отношении исходного сырья могут применяться другие требования, если они предусмотрены специальным законодательством. В определенных случаях применяются следующие положения:
a) для тканей (клеток), используемых для промышленного производства продукции (лекарственных средств), применяется соответствующее законодательство. Такие ткани и клетки становятся биологическими активными фармацевтическими субстанциями для некоторых видов биологических лекарственных препаратов (например, лекарственных препаратов тканевой инженерии, служащих для частичной или полной замены отдельных тканей), для которых применяются требования Правил надлежащей производственной практики Евразийского экономического союза и другие требования к лекарственным средствам;
b) в отношении высокотехнологичных лекарственных средств (ATMP), в которых кровь или компоненты крови используются в качестве исходного сырья, применяется соответствующее законодательство, которое устанавливает требования к отбору доноров, качеству и безопасности при заборе, тестировании, обработке, хранению и транспортированию человеческой крови и ее компонентов;
с) производство и контроль генетически модифицированных организмов должны соответствовать требованиям законодательства. Должна быть обеспечена и соблюдена соответствующая изоляция и другие меры защиты на объектах, где осуществляется какая-либо работа с генетически модифицированными микроорганизмами. Для установления и соблюдения необходимого уровня биологической безопасности должно быть получено соответствующее разрешение уполномоченных органов (организаций), если это предусмотрено законодательством, при этом также не должно быть противоречий с требованиями Правил надлежащей производственной практики Евразийского экономического союза.
Таблица
Руководство по производственной деятельности в рамках области
применения Требований к производству биологических (в том числе
иммунобиологических) активных фармацевтических субстанций и
лекарственных препаратов для медицинского применения
|
Тип и источник материалов |
Примеры продукции |
Применение Правил надлежащей производственной практики Евразийского экономического союза к стадиям производства1 |
|||
|
1. Животного или растительного происхождения: нетрансгенные |
гепарин, инсулин, ферменты, белки, экстракты аллергенов, высокотехнологичные лекарственные средства (ATMP), иммунные сыворотки |
сбор растений, забор органов, тканей или жидкостей2 |
измельчение, смешивание и (или) первичная обработка |
изоляция и очистка |
приготовление, наполнение |
|
2. Вирусы или бактерии (ферментация, культуры клеток) |
вирусные или бактериальные вакцины, ферменты, белки |
создание и поддержание главного3 и рабочего банков клеток, главного и рабочего вирусных посевных материалов |
культура клеток и (или) ферментация |
инактивация при необходимости, изоляция и очистка |
приготовление, наполнение |
|
3. Биотехнологи я (ферментация, культуры клеток) |
рекомбинантные продукты, моноклональные антитела, аллергены, вакцины, лекарственные препараты генной терапии (вирусные и невирусные векторы, плазмиды) |
создание и поддержание главного и рабочего банков клеток, главной и рабочей посевных культур |
культура клеток и (или) ферментация |
изоляция, очистка, модификация |
приготовление, наполнение |
|
4. Животного происхождения: трансгенные |
рекомбинантные белки, высокотехнологичные лекарственные средства (ATMP) |
главный и рабочий трансгенные банки |
измельчение, смешивание и(или) первичная обработка |
изоляция, очистка и модификация |
приготовление, наполнение |
|
5. Растительного происхождения: трансгенные |
рекомбинантные белки, вакцины, аллергены |
главный и рабочий трансгенные банки |
выращивание, сбор растений4 |
первичная, экстракция, изоляция, очистка, модификация |
приготовление, наполнение |
|
6. Человеческого происхождения |
ферменты, полученные из мочи, гормоны |
забор жидкостей 5 |
смешивание и (или) первичная обработка |
изоляция и очистка |
приготовление, наполнение |
|
7. Человеческого и (или) животного происхождения |
лекарственные препараты генной терапии: генетически модифицированные клетки |
донация, поставка и испытания исходных тканей (клеток)7 |
производство векторов6, очистка клеток и обработка Очистка и обработка производственных векторов6и клеток |
генетическая модификация клеток вне организма, создание главного и рабочего банков клеток или клеточного запаса |
приготовление, наполнение |
|
лекарственные препараты терапии соматическими клетками |
донация, поставка и испытания исходных тканей (клеток)7 |
создание главного и рабочего банков клеток или клеточного запаса |
изоляция клеток, очистка культур, соединение с неклеточными компонентами |
приготовление, соединение, наполнение |
|
|
лекарственные препараты тканевой инженерии |
донация, поставка и испытания исходных тканей (клеток) 7 |
первичная обработка, изоляция и очистка, создание главного и рабочего банков клеток, запаса первичных клеток |
изоляция клеток, очистка культур, соединение с неклеточными компонентами |
приготовление, соединение, наполнение |
|

1Серым отмечены стадии производства, к которым применяются требования Правил надлежащей производственной практики Евразийского экономического союза.
2Границы, в которых применяются принципы Правил надлежащей производственной практики Евразийского экономического союза, определяются в соответствии с разделом В1 настоящего приложения.
3Границы, в которых применяются принципы Правил надлежащей производственной практики Евразийского экономического союза, определяются в соответствии с разделом «Система посевной культуры и банка клеток».
4Для выращивания, сбора и первичной обработки, которые осуществляются в полевых условиях, может применяться руководство по правилам надлежащего выращивания и сбора исходного сырья растительного происхождения.
5 Применяются принципы Правил надлежащей производственной практики Евразийского экономического союза, в соответствии с разделом «Область применения».
6 В случае вирусных векторов, основной контроль соответствует контролю при производстве вирусов (строка 2 таблицы).
7 Ткани (клетки) человека должны соответствовать требованиям законодательства.
Принцип
Производство биологических активных фармацевтических субстанций и лекарственных препаратов имеет свою специфику, определяемую характером продукции и технологией производства. Характер производства, контроля и применения биологических лекарственных препаратов требует особых мер предосторожности.
В отличие от обычных лекарственных средств, производимых с использованием химических и физических методов, способных демонстрировать высокую степень надежности, производство биологических активных фармацевтических субстанций и лекарственных препаратов включает в себя использование биологических процессов и материалов, таких как культивирование клеток или экстрагирование материала из живых организмов. Эти биологические процессы могут демонстрировать свойственную им изменчивость, что приводит к существенному диапазону побочных продуктов различной природы. Поэтому принципы управления рисками для качества особенно важны для данного класса материалов и должны соблюдаться при разработке методов контроля на всех стадиях производства для минимизации вариабельности и уменьшения возможности контаминации и перекрестной контаминации.
Материалы, питательные среды и условия культивирования целевых микроорганизмов, культур клеток, вирусов и т. п. во многом способствуют возможности роста контаминирующих агентов. Кроме того, многие лекарственные препараты имеют ограниченную устойчивость к широкому спектру методов очистки, в особенности к методам, предназначенным для инактивации или устранения посторонних вирусных контаминантов. Для сведения к минимуму возможности такой контаминации основное внимание следует уделять планированию технологического процесса, конструкции оборудования, помещений, систем обеспечения, условиям приготовления и добавления буферов и реагентов, отбору проб и обучению персонала.
Спецификации на продукцию (в частности спецификации в фармакопейных статьях, общих фармакопейных статьях, регистрационное досье) должны определять допустимость определенного уровня бионагрузки (с указанием степени и стадии) для веществ и материалов, или требования к их стерильности. Производство также должно соответствовать другим требованиям, изложенным в регистрационном досье либо в протоколе клинического исследования (например, количество генераций (удвоений, пассажей) между посевной культурой или банком клеток).
Для биологических материалов, которые не могут быть простерилизованы (например, путем фильтрации), производство должно осуществляться в асептических условиях для минимизации риска внесения контаминантов. Для регламентации определенных производственных методов, например для удаления или инактивации вирусов, следует руководствоваться соответствующими процедурами. Применение соответствующих контроля и мониторинга за состоянием производственной среды и, где возможно, системы уборки и стерилизации «на месте» вместе с использованием закрытых систем может значительно уменьшить риск случайной контаминации и перекрестной контаминации.
Контроль обычно включает в себя биологические аналитические методы, которые характеризуются более высокой степенью вариабельности, чем физико-химические методы. Поэтому при производстве биологических активных фармацевтических субстанций и лекарственных препаратов ключевую роль играет надежный производственный процесс и особое значение имеет контроль в процессе производства.
Биологические лекарственные препараты, в состав которых входят ткани (клетки) донора (например, определенные высокотехнологичные лекарственные средства (ATMP)), должны соответствовать требованиям законодательства в части прослеживаемости, уведомления регуляторных органов о неблагоприятных реакциях и клинических случаях в ходе терапии, а также в части технических требований по идентификации, обработке, предохранению, хранению и транспортированию тканей (клеток) донора. Забор материалов и проведение испытаний должны проводиться в соответствии с системой качества, для которой определены стандарты и технические требования. Кроме того, требования нормативных правовых актов распространяются на прослеживаемость в отношении донора (с соблюдением конфиденциальности донора) через стадии, осуществляемые в учреждении по забору (проверке) тканей, и до учреждения, где используется лекарственный препарат (в соответствии с требованиями нормативных правовых актов).
Биологические активные фармацевтические субстанции и лекарственные препараты должны соответствовать требованиям действующих нормативных правовых актов в отношении уменьшения риска передачи возбудителя губчатой энцефалопатии животных и латентных вирусов через лекарственные препараты для медицинского применения и ветеринарные лекарственные препараты.
Часть А. Общее руководство
Персонал
1. Персонал, работающий в зонах производства и контроля биологических активных фармацевтических субстанций и лекарственных препаратов (в том числе персонал, занятый очисткой, обслуживанием или контролем качества), должен проходить обучение и периодическое повторное обучение в соответствии со своими обязанностями и спецификой производимой продукции, включая все особые меры предосторожности для защиты продукции, персонала и окружающей среды.
2. Для обеспечения безопасности продукции должно приниматься во внимание здоровье персонала. Сотрудники, занятые в производстве, техническом обслуживании, проведении испытаний и уходе за животными (в том числе контроле), при необходимости должны быть вакцинированы соответствующими специфическими вакцинами, а также проходить регулярные медицинские осмотры.
3. Любые заболевания персонала, которые могут неблагоприятно повлиять на качество продукта, должны препятствовать работе такого персонала в производственной зоне, а соответствующие записи сохраняться. В производстве вакцины БЦЖ и лекарственных препаратов туберкулина могут быть заняты только сотрудники, которые регулярно проходят проверку иммунного статуса или рентгенологическое обследование грудной клетки. Сотрудники должны проходить медицинский осмотр с учетом риска, которому они подвержены, медицинский осмотр требуется для персонала, работающего с опасными организмами.
4. С целью минимизации возможности перекрестной контаминации необходимо контролировать ограничение движения персонала (в том числе сотрудников службы контроля качества, специалистов по обслуживанию и уборке) на основе принципов управления рисками для качества. Как правило, не допускается переход сотрудников из зон, где возможен контакт с живыми микроорганизмами, генетически модифицированными организмами, токсинами или животными, в зоны, где проводятся работы с другой продукцией, инактивированной продукцией или другими организмами. Если подобных переходов избежать невозможно, должны быть приняты меры для контроля контаминации согласно принципам управления рисками для качества.
Помещения и оборудование
5. Являясь частью стратегии контроля, степень контроля производственной среды в отношении контаминации частицами и микроорганизмами в производственных помещениях должна соответствовать виду активной фармацевтической субстанции, промежуточной и готовой продукции и стадии технологического процесса. При этом необходимо учитывать уровень контаминации исходных материалов и степень риска для готового продукта. В программу мониторинга производственной среды должны быть дополнительно включены методы для определения присутствия специфических микроорганизмов (например, организма-хозяина, дрожжевых, плесневых грибов, анаэробных микроорганизмов и т. п.), если на это указывает процесс управления рисками для качества.
6. Производственные и складские помещения должны быть спроектированы с учетом требований к классам чистоты, а процессы спланированы таким образом, чтобы предотвратить контаминацию продукции посторонними веществами. Предотвращение контаминации является более эффективным, чем ее обнаружение и устранение, хотя контаминация, вероятно, будет проявляться во время таких производственных процессов, как ферментация и культивирование клеточных культур. Контрольные измерения, включая контроль систем обеспечения и контроль производственной среды, должны проводиться в соответствии с принципами управления рисками для качества на участках, где проходят открытые процессы и, соответственно, продукция может быть подвержена непосредственному воздействию производственной среды (например, во время добавления вспомогательных веществ, сред, буферов, газов, работ во время производства высокотехнологичных лекарственных средств (ATMP)). При выборе последовательных классов чистоты в производственных помещениях и соответствующих методов контроля принципы управления рисками для качества должны учитывать принципы, изложенные в соответствующих пунктах приложения № 1 к Правилам надлежащей производственной практики Евразийского экономического союза.
7. Работа с живыми клетками, устойчивыми к среде производственных помещений, должна осуществляться в специально предназначенных производственных помещениях. Если в производстве применяются патогенные микроорганизмы (например, 1 и 2 группы патогенности), то оно также должно осуществляться только в специально предназначенных для этого производственных помещениях.
8. Использование одного помещения для производства нескольких лекарственных препаратов может быть разрешено, если частью эффективной стратегии контроля, направленной на предотвращение перекрестной контаминации являются следующие или эквивалентные им факторы и мероприятия (соответственно рассматриваемым типам продукции):
a) знание основных характеристик всех клеток, организмов и любых посторонних агентов (например, патогенность, возможность обнаружения, устойчивость, восприимчивость к инактивации), работа с которыми осуществляется в одних и тех же помещениях;
b) при производстве продукции из многочисленных маленьких серий, получаемых из различного исходного сырья (например, лекарственные препараты на основе клеточных технологий), на стадии разработки стратегии контроля должны учитываться такие факторы, как состояние здоровья доноров, с целью снижения риска полной потери продукции;
c) предотвращение попадания живых микроорганизмов и спор в смежные помещения или на оборудование путем определения всех потенциальных маршрутов перекрестной контаминации и использования одноразовых компонентов и соответствующих инженерных мероприятий (например, закрытых систем);
d) наличие мероприятий контроля по удалению микроорганизмов и спор перед последующим производством другой продукции. Процедуры очистки и деконтаминации от микроорганизмов и спор должны быть отвалидированы (в том числе и для систем отопления, вентиляции и кондиционирования воздуха (HVAC));
e) если микроорганизмы являются устойчивыми к условиям производственной среды и в распоряжении имеются соответствующие методы, контроль производственной среды, специфический для получаемого микроорганизма, следует проводить в смежных зонах во время производства и после завершения очистки и деконтаминации. Также нужно принимать во внимание риски, связанные с использованием определенного контрольно-измерительного оборудования (например, для определения частиц в воздухе) в зонах, где проводятся работы с живыми и (или) спорообразующими микроорганизмами;
f) продукция, оборудование, вспомогательное оборудование (например, для калибровки и валидации) и одноразовые материалы должны перемещаться в пределах предназначенных зон и должны удаляться из этих зон таким образом, чтобы предотвратить контаминацию других зон, другой продукции на различных стадиях производства (например, следует предотвратить контаминацию инактивированных продуктов или анатоксинов не инактивированными продуктами);
g) производство на основе принципа проведения однотипных циклов производства (кампаний).
9. Необходимость наличия специализированных помещений для финишной обработки (например, для приготовления, наполнения, упаковки) будет зависеть от перечисленных в пункте 8 настоящего Приложения факторов, а также от дополнительных факторов в отношении специфики биологического лекарственного препарата и характеристик другой продукции, включая любые небиологические продукты, производимые в тех же помещениях. На заключительных стадиях могут потребоваться другие меры контроля определенной последовательности добавления веществ, скорости перемешивания, времени и температуры, предельного времени воздействия света и герметизации (изоляции), а также в процедурах очистки в случае пролива (рассыпания).
10. Мероприятия и процедуры, необходимые для обеспечения безопасности производственной среды и персонала, не должны противоречить мероприятиям и процедурам, необходимым для обеспечения качества продукта.
11. Системы воздухоподготовки должны быть спроектированы, сконструированы и обслуживаться таким образом, чтобы исключить риск перекрестной контаминации между различными производственными зонами. Также может возникнуть необходимость в отдельных системах воздухоподготовки для определенных зон. Решение относительно использования систем вентиляции без рециркуляции должно быть принято на основании принципов управления рисками для качества.
12. Работу со стерильной продукцией необходимо осуществлять в зонах с избыточным давлением, но в особых зонах в точках локализации патогенных микроорганизмов следует создавать отрицательный перепад давления для предотвращения распространения контаминантов за пределы этих зон. Если для работы в асептических условиях с материалами, представляющими особый риск (например, с патогенными микроорганизмами), используются зоны с пониженным давлением или безопасные боксы, их следует окружать зонами соответствующего класса чистоты с избыточным давлением. Данные перепады давления должны быть четко определены и находиться под постоянным контролем с соответствующими настройками аварийной сигнализации.
13. Конструкция оборудования, используемого для работы с живыми микроорганизмами и клетками, включая оборудование для отбора проб, должна исключать возможность контаминации во время проведения работ.
14. Конструкция, обеспечивающая первичную изоляцию, должна исключать риск утечки биологических агентов в непосредственное рабочее пространство, что должно быть подтверждено результатами тестирования с определенной периодичностью.
15. Рекомендуется по возможности использовать системы «очистки на месте» и «обработки паром на месте» («стерилизации на месте»). Конструкция вентилей на ферментаторах должна предусматривать возможность их стерилизации паром.
16. Воздушные фильтры должны быть гидрофобными, срок их службы должен быть определен в процессе валидации путем проверки целостности с определенной периодичностью согласно соответствующим принципам управления рисками для качества.
17. Конструкция дренажных систем должна позволять проводить эффективную нейтрализацию и деконтаминацию сточных вод для исключения риска перекрестной контаминации. Должно быть обеспечено выполнение требований законодательства с целью минимизации рисков контаминации окружающей среды в соответствии с рисками, связанными с биологической опасностью отходов производства.
18. Ввиду изменчивости свойств биологических лекарственных средств или процессов их производства необходимо отмеривать или взвешивать какое-либо соответствующее (критическое) исходное сырье (например, питательные среды и буферы) в ходе технологического процесса. В этих случаях в производственной зоне допускается хранение небольших запасов этого исходного сырья на протяжении срока, установленного на основании соответствующих критериев (например, длительность производства серии или длительность кампании).
Животные
19. Для производства биологических лекарственных препаратов используются разные виды животных. Выделяются следующие 2 большие группы источников:
a) живые животные, объединенные в группы, стада, стаи. Например, обезьяны (вакцина против полиомиелита), лошади, овцы и козы (иммунные сыворотки против ядов змей и столбняка), кошки (аллергены), кролики, мыши и хомяки (вакцина против бешенства), козы, крупный рогатый скот (трансгенные продукты);
b) ткани (клетки) животного происхождения, извлеченные из умерщвленных животных (овцы и свиньи), используемые для производства высокотехнологичных лекарственных средств (ATMP) или как сырье для ферментов, антикоагулянтов и гормонов.
Животные также используются для контроля качества в соответствии со спецификациями (пирогенность, токсичность, безвредность, специфическая активность).
20. В дополнение к выполнению нормативных требований в отношении губчатой энцефалопатии животных другие опасные агенты (возбудители зоонозов, антропозоонозов животных) должны контролироваться и регистрироваться согласно постоянно действующей программе мониторинга. Необходима консультация соответствующего специалиста для организации таких программ.
В случаях заболевания животных-доноров или животных, которые используются в качестве сырья, должны быть проведены и зарегистрированы соответствующие исследования на предмет пригодности этих животных и пригодности животных, пребывавших в контакте с больным животным, для использования при производстве (в качестве исходных материалов или исходного сырья), контроле качества продукции и проведении испытаний на безопасность. Должна быть принята в форме письменного документа процедура ретроспективного анализа, позволяющая принимать решение относительно годности биологической активной фармацевтической субстанции или лекарственного препарата, в состав которого входит или при производстве которого использовался такой животный материал в качестве исходного сырья или материала. Для установления того, какая из донаций последней показала отрицательный результат на наличие заболевания, если применимо, проводится повторное тестирование образцов, сохраненных при предыдущем заборе материала от того же животного-донора (если таковые имеются), что влияет на процесс принятия данного решения. При определении периода изъятия животных из программы должен учитываться период выведения терапевтических агентов, использовавшихся для введения животным-донорам или животным, которые использовались в качестве сырья, что должно быть оформлено документально.
21. Особое внимание следует уделять предотвращению и мониторингу инфекционных заболеваний у животных, которые используются в качестве сырья, и у животных-доноров. Принимаемые меры должны включать контроль источников, помещений, пастбища, контроль биологической безопасности, режимов проведения испытаний, контроль вспомогательных материалов и кормов для животных. Особенное значение данные проверки имеют для животных, свободных от специфических патогенов, в соответствии с требованиями нормативных актов. Должны быть определены требования к содержанию и мониторингу здоровья других животных (например, живущих в стаях или стадах).
22. Для лекарственных препаратов, произведенных с использованием трансгенных животных, должна быть обеспечена прослеживаемость исходных животных, использованных для создания трансгенных животных.
23. Особое внимание должно быть уделено нормативным актам в отношении защиты животных, используемых в экспериментальных целях, в отношении вивариев, в которых содержатся животные, ухода за животными и карантина. Виварии, в которых содержатся животные, используемые для производства и контроля качества биологических активных фармацевтических субстанций и лекарственных препаратов, должны быть отделены от зон производства и контроля качества.
24. Для различных видов животных должны быть определены основные показатели, которые затем должны контролироваться и регистрироваться. Показатели могут включать возраст, пол, вес и состояние здоровья животных.
25. Для предотвращения риска перепутывания и возможных опасностей, должна существовать система идентификации в отношении животных, биологических агентов и проведенных испытаний.
Документация
26. Для исходных сырья и материалов для производства биологических лекарственных препаратов может потребоваться дополнительная информация об источнике, происхождении, цепи поставки, методе производства и применяемых методах контроля качества для обеспечения необходимого объема контроля, в том числе микробиологического контроля.
27. Для некоторых типов продукции может потребоваться специфическое описание материалов, входящих в серию, в частности, соматических клеток, используемых при производстве высокотехнологичных лекарственных средств (ATMP). В случаях аутологичного лекарственного препарата и лекарственного препарата от специально подобранного донора продукция должна рассматриваться как одна серия.
28. Для лекарственных препаратов, при производстве которых используются клетки человека или ткани доноров, должна быть обеспечена полная прослеживаемость, начиная от исходного сырья и материалов, в том числе содержащая информацию обо всех веществах, контактировавших с клетками или тканями, вплоть до подтверждения получения лекарственного препарата в месте его применения. В то же время должна быть обеспечена анонимность пациентов и конфиденциальность информации об их здоровье. Соответствующие записи, обеспечивающие прослеживаемость лекарственного препарата, должны быть сохранены на протяжении тридцати лет после даты окончания срока годности лекарственного препарата. Особое внимание должно уделяться обеспечению прослеживаемости специфических лекарственных препаратов для использования в особых случаях (например клетки от специально подобранного донора). Производство лекарственных средств с использованием компонентов крови в качестве исходного сырья или материалов должно соответствовать требованиям законодательства. В соответствии с законодательством при производстве высокотехнологичных лекарственных средств (ATMP) должна быть обеспечена прослеживаемость клеток человека, в том числе гемопоэтических стволовых клеток. Мероприятия, обеспечивающие прослеживаемость и хранение документации в течение необходимого периода, должны быть включены в технические соглашения между вовлеченными в такую деятельность сторонами.
Производство
29. Учитывая возможную высокую изменчивость свойств биологических веществ и продуктов, необходимо обеспечить на разных этапах жизненного цикла продукции (например, на этапе разработки процесса) повышение надежности и устойчивости процесса, снижая тем самым его вариабельность и повышая воспроизводимость.
Повторная оценка должна проводиться в процессе обзоров качества продукции.
30. Поскольку среды и реактивы в условиях культивирования обеспечивают рост клеток или микроорганизмов, которые, как правило, являются монокультурами, особое внимание должно уделяться стратегии контроля качества для гарантии предупреждения и минимизации бионагрузки и связанного с ней загрязнения метаболитами и эндотоксинами. Для высокотехнологичных лекарственных средств (ATMP) на основе клеток, которые, как правило, производятся малыми сериями, риск перекрестной контаминации между клеточными лекарственными препаратами от разных доноров с различным состоянием здоровья должен контролироваться согласно установленным требованиям и процедурам.
Исходное сырье и материалы
31. Следует четко определять источник, происхождение и пригодность биологических исходных сырья и материалов (например, криопротекторов, питающий клеток, реагентов, питательных сред, буферов, сывороток, ферментов, цитокинов, факторов роста) для последующего их использования. Если проведение необходимых испытаний занимает много времени, допускается начинать обработку исходного сырья до получения результатов этих испытаний, но для использования этих исходных материалов должно быть учтено их влияние на другие серии в случае выявления несоответствия и оценены риски согласно принципам управления рисками для качества. В таких случаях выдача разрешения на выпуск серии готовой продукции зависит от удовлетворительных результатов испытаний исходного сырья. На основании требований к соответствующим стадиям производства должна проводиться идентификация всех исходных материалов. Дополнительные руководящие указания для биологических лекарственных препаратов приведены в части I Правил надлежащей производственной практики Евразийского экономического союза и приложении № 2 к Правилам надлежащей производственной практики Евразийского экономического союза, а для биологических активных фармацевтических субстанций в части II Правил надлежащей производственной практики Евразийского экономического союза.
32. При оценке рисков контаминации исходных сырья и материалов во время их прохождения по цепи поставки особое внимание следует уделять риску, связанному с губчатой энцефалопатией животных. Также должно быть уделено внимание материалам, непосредственно контактирующим с технологическим оборудованием или продукцией (например, питательным средам, используемым для моделирования асептического процесса, и смазочным материалам, которые могут контактировать с продуктом).
33. Вследствие того, что риски внесения контаминации и соответствующие последствия для готового лекарственного препарата не зависят от стадии производства, установление стратегии контроля для защиты продукции, приготовления растворов, буферов и добавляемых компонентов должно основываться на принципах и руководящих указаниях, содержащихся в соответствующих пунктах приложения № 1 к Правилам надлежащей производственной практики Евразийского экономического союза. Особую важность имеют контрольные мероприятия, необходимые для проверки качества исходных материалов, а также для процесса асептического производства продукции на клеточной основе, где финишная стерилизация не является возможной, а способность удаления микробных побочных продуктов ограничена. Когда в регистрационном досье или протоколе клинических исследований установлен допустимый тип и уровень бионагрузки (например, на стадии получения активной фармацевтической субстанции), стратегия контроля должна предусматривать способы, которыми будет поддерживаться установленный уровень бионагрузки.
34. При необходимости стерилизации исходного сырья и материалов она по возможности должна проводиться термическим методом. При необходимости могут также использоваться другие соответствующие методы, использующиеся для инактивации биологических материалов (например, радиация и фильтрация).
35. Может потребоваться проведение других мероприятий, в частности, использование антибиотиков на ранних этапах производства с целью уменьшения бионагрузки, которая возможна при поставке живых тканей (клеток). Следует по возможности избегать таких мероприятий, но в случае необходимости их использование должно быть обосновано, а их применение в технологическом процессе должно быть прекращено на стадии, указанной в регистрационном досье или протоколе клинических исследований.
36. Для тканей (клеток) человека, используемых в качестве исходных материалов для биологических лекарственных средств, необходимо учитывать следующие требования:
a) приобретение, донация и проведение испытаний должно регулироваться законодательством государств - членов Евразийского экономического союза (далее - государства-члены). Учреждения, поставляющие исходное сырье, должны получать разрешение уполномоченных органов государств-членов в соответствии с законодательством государств-членов. Наличие необходимых разрешений должно быть проверено в рамках системы управления поставками;
b) в случаях импортирования тканей (клеток) человека из других стран, должны быть соблюдены соответствующие стандарты контроля качества и безопасности, эквивалентные нормативным правовым актам государств-членов. Также должны быть соблюдены установленные законодательством государств-членов требования прослеживаемости и сообщений о серьезных нежелательных реакциях и серьезных нежелательных явлениях;
c) в некоторых случаях работа с тканями (клетками), используемыми в качестве исходных материалов для биологических лекарственных препаратов, производится в учреждениях по забору (проверке) тканей (например, для создания начальных клеточных банков или клеточных линий, предшествующих созданию главного банка клеток). В этих случаях, в соответствии с законодательством, должно быть ответственное лицо, которое отвечает за эти этапы работы;
d) ответственное лицо в учреждении по забору (проверке) тканей выдает разрешение на использование тканей (клеток) перед их поставкой производителю лекарственного препарата, после чего применяются стандартные процедуры контроля исходных материалов. Результаты испытаний всех тканей (клеток), поставляемых учреждением по забору (проверке) тканей, должны быть представлены производителю лекарственного препарата. Данная информация должна использоваться для соответствующего разделения материалов и определения способов хранения. В случае необходимости возможна доставка тканей (клеток) производителю лекарственного препарата до получения результатов испытаний от учреждения по забору (проверке) тканей. Это возможно при наличии соответствующих мер контроля для предотвращения перекрестной контаминации тканями (клетками), на что было получено разрешение ответственного лица в учреждении по забору (проверке) тканей;
e) транспортирование тканей (клеток) человека к производственной площадке должно осуществляться в соответствии с письменным соглашением между ответственными сторонами. Производственные площадки должны иметь документальное подтверждение соблюдения соответствующих специфических условий хранения и транспортирования;
f) должны соблюдаться требования прослеживаемости, начиная с учреждения по забору (проверке) тканей до доставки получателю, включая материалы, находившиеся в контакте с тканями (клетками);
g) должно существовать соглашение между ответственными сторонами (например, производителями, учреждениями по забору (проверке) тканей, спонсорами, держателями регистрационных удостоверений), которое определяет сферы ответственности каждой из сторон (включая указание ответственных и Уполномоченных лиц).
37. В отношении генной терапии необходимо учитывать следующие требования:
a) для продукции, произведенной с использованием вирусных векторов, исходные материалы являются компонентами, из которых получен вирусный вектор, то есть главный вирусный посевной материал или плазмиды, которые используются для трансфекции пакующих клеток, и главного банка клеток, используемого для линии пакующих клеток;
b) для продукции, произведенной с использованием плазмид, невирусных векторов и генетически модифицированных микроорганизмов, за исключением вирусов и вирусных векторов, исходными материалами являются компоненты, используемые для создания клеток-продуцентов, то есть плазмиды, бактерия-хозяин и главный банк рекомбинантных микроорганизмов;
c) для генетически модифицированных клеток исходными материалами являются компоненты, используемые для получения генетически модифицированных клеток, то есть исходные материалы для производства векторов, а также клетки человека или животных;
d) принципы настоящих Правил применяются, начиная от системы банка клеток, используемого для производства вектора или плазмиды, которые используются для переноса генов.
38. При производственных процессах, в которых клетки человеческого или животного происхождения используются в качестве питающих клеток, должен проводиться соответствующий контроль источников, испытаний, транспортирования и хранения данных материалов, включая контроль в соответствии с требованиями законодательства.
Система посевной культуры и банка клеток
39. Для предотвращения нежелательного изменения свойств, которое может произойти вследствие многократных пересевов или большого числа генераций, производство биологических активных фармацевтических субстанций и лекарственных препаратов, получаемых из культур микроорганизмов, культур клеток или размножением в эмбрионах, тканях и органах животных, должно быть основано на системе главной и рабочей вирусных посевных культур и (или) банков клеток. Такая система может быть неприменимой ко всем типам высокотехнологичных лекарственных средств (ATMP).
40. Количество генераций (удвоений, пассажей) между посевной культурой или банком клеток и биологической активной фармацевтической субстанцией и лекарственным препаратом должно соответствовать требованиям спецификаций в регистрационном досье или протоколе клинических исследований.
41. Создание систем посевных культур и банков клеток, включая главные и рабочие посевные культуры, должно являться частью управления жизненным циклом продукции и проводиться в соответствующих условиях. Производственная среда должна находиться под соответствующим контролем для обеспечения безопасности систем посевных культур и банков клеток, а также персонала, работающего с ними. При создании посевных культур и банков клеток не допускается одновременная работа с другими живыми или инфицирующими материалами (например, вирусами, линиями или штаммами клеток) в одной и той же зоне или одного и того же персонала. Должна быть доступна документация, позволяющая обеспечить прослеживаемость стадий, предшествующих генерации главной посевной культуры или главного банка клеток, где могут быть применены только принципы настоящих Правил. Данная документация должна включать информацию относительно компонентов, использовавшихся во время разработки и возможно влияющих на безопасность продукции (например, реагенты биологического происхождения), от начального источника и до получения генно-инженерного продукта, если применимо.
42. После формирования главного и рабочего банков клеток и главной и рабочей посевных культур должны соблюдаться процедуры по карантину и разрешению к использованию. Должны быть проведены соответствующие квалификация и испытания в отношении контаминантов. Их дальнейшая пригодность впоследствии должна быть подтверждена стабильностью характеристик и качеством последующих серий продукции. Доказательство стабильности и воспроизводимости посевных культур или банков клеток необходимо оформлять документально. Содержание записей должно позволять проводить оценку тенденций (трендов).
43. Посевные культуры и банки клеток следует создавать, хранить и использовать таким образом, чтобы риск их контаминации или изменения был минимальным (например, хранить в герметичных контейнерах в жидком азоте). В случае хранения различных посевных культур и банков клеток в одних и тех же зонах или с использованием одного и того же оборудования должны быть приняты меры по предотвращению перепутывания и перекрестной контаминации с учетом инфекционной природы материалов.
44. Лекарственные препараты на основе клеток зачастую производятся из клеточного запаса, полученного из ограниченного количества пассажей. В отличие от двухуровневой системы главных и рабочих банков клеток, количество производственных циклов на основе клеточного запаса ограничено количеством аликвот, полученных после роста, и не распространяется на весь жизненный цикл продукции. Протокол валидации должен учитывать изменения клеточного запаса.
45. Емкости для хранения должны быть герметично закрыты, четко маркированы; содержаться при соответствующей температуре. Необходимо вести документальный учет хранящихся емкостей. Температуру хранения следует непрерывно регистрировать, а в установках с жидким азотом контролировать его уровень. Отклонения параметров хранения от установленных пределов и любые предпринятые корректирующие и предупреждающие действия должны быть оформлены документально.
46. Рекомендуется разделять запасы на части и хранить раздельно во избежание полной утраты. Контроль месторасположения должен обеспечивать выполнение указанных требований.
47. Условия хранения и обработки запасов должны определяться согласно тем же самым процедурам и параметрам. После взятия контейнеров из хранилища посевной культуры (банка клеток) не допускается возвращать их в хранилище повторно.
Принципы работы
48. При управлении изменениями с установленной периодичностью должны рассматриваться эффекты, включая кумулятивные эффекты изменений (например, в производственных процессах), влияющие на качество, безопасность и эффективность готового лекарственного препарата.
49. Критические операционные (технологические) или другие исходные параметры, влияющие на качество лекарственного препарата, должны быть определены, валидированы, документированы и поддерживаться в соответствии с установленными требованиями.
50. Стратегия контроля поступления сырья и материалов в производственные зоны должна основываться на принципах управления рисками для качества. Для асептических процессов термостойкие сырье и материалы, попадающие в чистую или чистую и изолированную зону должны, по возможности, поступать в них через проходной автоклав или сухожаровой шкаф. Нетермостойкие сырье и материалы должны вноситься через воздушные шлюзы с блокировкой дверей, подвергаясь процедурам эффективной санитарной обработки поверхности. Разрешается стерилизация предметов и материалов в другом месте при условии, что они содержат количество оберток, соответствующих числу стадий, необходимых для прохождения в чистую зону, и вносятся в нее через воздушный шлюз с соблюдением соответствующих мер предосторожности путем санитарной обработки поверхности.
51. Должны быть подтверждены ростовые свойства питательных сред с целью доказательства их пригодности для предполагаемого использования. Питательные среды должны, по возможности, стерилизоваться на месте. При плановой подаче газов, питательных сред, кислот или щелочей, пеногасителей и т. п. к ферментерам по возможности следует использовать стерилизующие фильтры, встроенные в линии подачи.
52. Добавление веществ или культур в ферментеры и другие сосуды, а также отбор проб из них необходимо проводить в тщательно контролируемых условиях для предотвращения контаминации. При внесении добавок или отборе проб необходимо контролировать правильность подсоединения сосудов.
53. При необходимости следует вести постоянный контроль некоторых производственных процессов (например, ферментации) с внесением результатов контроля в записи по производству серии. При производстве с использованием метода непрерывного культивирования следует обратить внимание на специальные требования к контролю качества, возникающие в результате выбора такого производственного метода.
54. Процессы центрифугирования и смешивания продуктов могут привести к образованию аэрозолей, поэтому во избежание перекрестной контаминации эти процессы следует проводить в изолированных зонах.
55. При случайной утечке, в особенности живых микроорганизмов, должны быть приняты неотложные меры безопасности. Для каждого вида или группы микроорганизмов должны быть предусмотрены специальные мероприятия по деконтаминации. При использовании различных штаммов бактерий одного вида или очень похожих вирусов эта процедура может быть валидирована в отношении только одного штамма или вируса, при отсутствии существенных различий в устойчивости к соответствующему агенту (агентам) для деконтаминации.
56. Если материалы, используемые для производства и контроля, а также бумажные носители информации явно являются контаминированными (например, пролитыми жидкостями, аэрозолями или потенциально опасными микроорганизмами), они должны быть соответственно дезинфицированы или информация на бумажных носителях информации должна передаваться иными способами.
57. При инактивации или удалении вирусов в ходе производства необходимо принимать меры против повторной контаминации обработанной продукции со стороны необработанной продукции.
58. Для продуктов, инактивируемых при помощи добавления реагентов (например, микроорганизмы в процессе производства вакцин), процесс должен гарантировать полную инактивацию живых микроорганизмов. После тщательного смешивания культуры и инактивирующего агента должны учитываться все контактирующие с продуктом поверхности, пребывавшие в контакте с культурой.
59. При применении хроматографических методов используются разные виды оборудования. Принципы управления рисками для качества должны соблюдаться при разработке стратегии контроля сорбентов, корпусов колонок и другого оборудования при их использовании для производства в режиме кампаний или в помещениях для производства нескольких лекарственных препаратов. Не рекомендуется использование одних и тех же сорбентов на разных технологических стадиях. Следует установить критерии приемлемости, условия работы, методы восстановления, срок службы и методы стерилизации или дезинфекции колонок.
60. Дополнительные инструкции относительно использования облученных оборудования и материалов приведены в приложении № 12 к Правилам надлежащей производственной практики Евразийского экономического союза.
61. Должна существовать система, гарантирующая целостность и герметичность контейнеров после их наполнения, и должны быть предусмотрены процедуры на случай любых утечек или просыпаний (проливов) в случае, если продукт или промежуточный продукт представляют особый риск. Для операций по розливу и упаковке должны существовать процедуры по соблюдению условий, обеспечивающих поддержание продукта в установленных пределах (например, время и (или) температура).
62. Работа с контейнерами (ампулами, флаконами и др.), содержащими биологические агенты, должна проводиться таким образом, чтобы избежать контаминации других лекарственных препаратов или проникновения живых агентов в производственную или окружающую среду. Для принятия решения относительно управления указанными рисками должны быть приняты во внимание жизнеспособность таких организмов и их биологическая классификация (группа патогенности).
63. Следует уделять должное внимание подготовке, печати, хранению и нанесению этикеток на упаковку, в том числе нанесению на первичную (внутренюю) и вторичную (потребительскую) упаковку специфической информации для пациент-специфических препаратов (продуктов) или об использовании методов генетической инженерии. В случае если высокотехнологичные лекарственные средства (ATMP) предназначены для аутологичного применения, на этикетке должны указываться уникальный идентификатор пациента и надпись: «Только для аутологичного применения». Если вторичная (потребительская) упаковка отсутствует, данная информация должна указываться на первичной (внутренней) упаковке.
64. В случае использования сверхнизких температур хранения должна быть подтверждена устойчивость маркировки к используемым температурам.
65. Когда информация о состоянии здоровья донора (человека или животного), имеющая значение для качества продукции, становится доступной после закупки, это должно учитываться в процедурах отзыва.
Контроль качества
66. Контроль в процессе производства является более важным для обеспечения стабильности качества биологических активных фармацевтических субстанций и лекарственных препаратов, чем для других лекарственных препаратов. Межоперационный контроль должен осуществляться на соответствующих стадиях производства с целью контроля условий, являющихся важными для качества готового продукта.
67. В случаях, когда промежуточные продукты могут храниться длительное время (дни, недели или дольше), должна быть рассмотрена возможность включения в текущую программу испытания стабильности серий готовой продукции, которые произведены из промежуточных продуктов с максимальным периодом хранения в процессе производства.
68. Для определенных типов клеток (например, аутологичные клетки, используемые в производстве высокотехнологичных лекарственных средств (ATMP)), которые могут быть доступными в ограниченных количествах и в случае, если это допускается регистрационным досье, проведение испытаний и порядок хранения контрольных образцов могут быть изменены, что оформляется документально.
69. В отношении клеточных высокотехнологичных лекарственных средств тесты на стерильность должны проводиться на культурах клеток или банках клеток, свободных от антибиотиков, для получения доказательства отсутствия контаминации бактериями и грибами, а также для возможности обнаружения организмов, требующих специальных условий культивирования (при необходимости).
70. Соответствующая стратегия контроля должна осуществляться для производства биологических лекарственных препаратов с коротким сроком годности (до 14 дней), для которых требуется выпуск серии еще до окончания проведения испытаний качества всей партии готовой продукции (например, исследование стерильности). Такой контроль должен быть основан на глубоком понимании свойств лекарственного препарата и производственного процесса и должен принимать во внимание контроль и характерные свойства исходного сырья и материалов. Необходимо наличие четкого и полного описания всей процедуры выпуска, включающего перечень обязанностей отдельных сотрудников, вовлеченных в оценку производственных и аналитических данных. Должна проводиться непрерывная оценка эффективности системы обеспечения качества, включая ведение записей, позволяющих оценивать тенденции (тренды). Должны быть предусмотрены альтернативные методы (например, быстрые микробиологические методы) получения соответствующих результатов, позволяющих проводить предварительное подтверждение соответствия серий в случаях, если невозможно провести испытания готового лекарственного препарата из-за его короткого срока годности. Процедура подтверждения соответствия и выпуска серии может проводиться путем осуществления двух и более стадий:
a) оценка ответственным лицом записей, касающихся процесса производства серии, и результатов мониторинга производственной среды (при необходимости), которые должны включать условия производства, все отклонения от стандартных процедур и существующие аналитические результаты для первичного разрешения серии продукта к выпуску Уполномоченным лицом;
b) оценка Уполномоченным лицом результатов окончательных аналитических испытаний и другой доступной информации для заключительного подтверждения соответствия серии установленным требованиям.
Должна быть предусмотрена процедура, описывающая необходимые мероприятия (включая взаимодействие с медицинскими работниками), в случае получения результатов испытаний, выходящих за границы спецификаций. Такие случаи должны расследоваться в полном объеме. Соответствующие корректирующие и предупреждающие действия, направленные на предотвращение возможности повторения таких случаев, должны регистрироваться в форме письменного документа.
Часть B. Специальное руководство по отдельным типам продукции
B1. Лекарственные препараты животного происхождения
Настоящая часть применяется в отношении материалов животного происхождения, в том числе материалов, полученных из таких учреждений, как скотобойни. Поскольку цепи поставки могут быть обширными и сложными, должны применяться средства контроля, основанные на принципах управления рисками для качества. При этом необходимо учитывать фармакопейные требования, включая проведение соответствующих испытаний на определенных стадиях. Должна вестись соответствующая документация, обеспечивающая прослеживаемость цепи поставки с четким указанием роли каждого участника цепи поставки, включая, как правило, достаточно подробное описание схемы поставок.
1. Необходимо наличие программ мониторинга опасных для человека болезней животных (ветеринарное освидетельствование). При оценке факторов риска должны приниматься во внимание сообщения от заслуживающих доверие источников относительно распространенности заболевания на территории государства. Одной из организаций, осуществляющих мониторинг заболеваемости животных в мире, является Международное эпизоотическое бюро. Сообщения должны сопровождаться информацией о проверке состояния здоровья животных и программе (программах) контроля на государственном и местном уровнях. Последнее включает в себя мероприятия по контролю источников (например, фермы или загоны для скота), из которых получены животные, и контролю во время транспортирования животных на скотобойню.
2. Скотобойни должны соответствовать требованиям законодательства при использовании их в качестве поставщиков тканей животных. Должны быть приняты во внимание отчеты уполномоченных органов, подтверждающие соблюдение требований безопасности и качества кормов, и соответствие законодательству относительно животных и растений государств-членов и других стран, из которых сырье экспортируется на территорию государств-членов.
3. Мероприятия по контролю исходного сырья и материалов в таких организациях, как скотобойни, должны включать в себя определенные элементы системы управления качеством для обеспечения удовлетворительного уровня профессиональной подготовки персонала, прослеживаемости материалов, контроля и стабильности. Могут применяться меры, не предусмотренные законодательством при условии, что они обеспечивают соответствующий уровень контроля.
4. Должны быть предусмотрены мероприятия по контролю исходного сырья и материалов, обеспечивающие предотвращение вмешательств, влияющих на качество материалов, или, по меньшей мере, предоставляющие информацию о проведении таких мероприятий при продвижении исходных материалов или сырья по производственной цепочке или цепочке поставки. Указанные мероприятия должны проводиться в отношении перемещения материалов от мест первичного сбора, проведения частичной и полной очистки до мест хранения, накопления, размещения и нахождения у посредников. Следует вести детальную регистрацию проведенных мероприятий в рамках системы, обеспечивающей прослеживаемость продукции, включая регистрацию любых нарушений, связанных с ними расследований и принятых мер.
5. Должны проводиться постоянные аудиты поставщиков исходного сырья и материалов, подтверждающие соблюдение требований контроля материалов на разных стадиях производства. В наличии должна быть полная документация о расследованиях происшествий, проведенных с тщательностью, соответствующей значимости происшествий. Должны существовать системы, обеспечивающие проведение эффективных корректирующих и предупреждающих действий.
6. Ткани (клетки) и органы, используемые для производства ксеногенных клеточных лекарственных препаратов, должны быть получены исключительно от животных, которые содержатся в неволе изолированно от других животных и выращены специально для этих целей. Не допускается использование тканей (клеток) и органов диких животных или животных со скотобоен, а также тканей животных- основателей (животный организм, несущий чужеродный ген). Следует вести наблюдение и документацию относительно состояния здоровья животных.
7. При ксеногенной клеточной терапии должны соблюдаться соответствующие рекомендации относительно поставки и испытаний клеток животных. Требования относительно ксеногенных лекарственных препаратов предусматриваются соответствующими актами.
B2. Лекарственные препараты аллергенов
Материалы могут быть произведены путем извлечения из естественных источников или с использованием технологии рекомбинантной ДНК:
1. Для гарантии соответствия поставки исходных материалов должно существовать их описание, включающее в себя необходимые сведения (например, общепринятое и научное название, происхождение, природа, пределы содержания контаминантов, метод забора таких материалов). Животные материалы должны быть получены от здоровых животных. Для колоний (например, клещей, животных), которые используются для экстракции аллергенов, должна существовать соответствующая система контроля, обеспечивающая биологическую безопасность. Лекарственные препараты аллергенов должны храниться в соответствующих условиях, обеспечивающих их качество.
2. Стадии технологического процесса, включающие в себя предварительную обработку, экстракцию, фильтрацию, диализ, концентрирование или лиофилизацию, должны быть детально описаны и валидированы.
3. Процессы модификации, используемые для производства модифицированных экстрактов аллергенов (например, аллергоидов, конъюгатов), должны быть описаны в соответствующей документации. Промежуточные продукты в технологическом процессе должны быть идентифицированы и проконтролированы.
4. Смеси экстрактов аллергенов должны быть приготовлены из отдельных экстрактов исходных материалов, полученных из одного источника. Каждый отдельный экстракт должен быть определен как отдельная активная фармацевтическая субстанция.
B3. Лекарственные препараты иммунных сывороток животных
1. Особое внимание должно уделяться контролю антигенов биологического происхождения для гарантии их качества, постоянства и отсутствия побочных агентов. Подготовка материалов, используемых для иммунизации животных (например, использование (введение) антигенов, гаптен-носителей, адъювантов, стабилизирующих агентов), и хранение таких материалов непосредственно перед иммунизацией должны производиться в соответствии с процедурами, принятыми в форме письменного документа.
2. Процедуры иммунизации, исследования крови и забора крови должны проводиться в соответствии с регистрационным досье.
3. Условия производства лекарственных препаратов из субфрагментов антител (например, участки связывания антигена Fab и F(ab’)2) и любые дальнейшие модификации должны соответствовать валидированным и утвержденным параметрам. Если ферменты, используемые при производстве, состоят из нескольких компонентов, должна быть обеспечена их стабильность.
B4. Вакцины
1. При использовании эмбрионов птиц должно быть обеспечено здоровье всех стай, используемых для их получения (для стай, свободных от специфических патогенов, и для здоровых стай).
2. Должна проводиться валидация целостности контейнеров, используемых для хранения промежуточных продуктов, и времени их хранения.
3. В зонах, содержащих живые биологические агенты, запрещается открывание сосудов, содержащих инактивированные лекарственные препараты, и отбор проб из них.
4. Последовательность прибавления активных компонентов, адъювантов и вспомогательных веществ в процессе производства промежуточного или готового продукта должна соответствовать технологическим инструкциям.
5. В случае использования для производства или испытаний микроорганизмов, которым присвоен высший уровень биологической опасности (например, пандемические штаммы), должны быть обеспечены необходимые меры изоляции. Должно быть получено документальное подтверждение разрешения на проведение указанных мероприятий от соответствующего уполномоченного органа. Указанная документация должна быть принята в форме письменного документа и доступна для проверки.
B5. Рекомбинантные продукты
1. Для обеспечения постоянства свойств лекарственного препарата, содержащего допустимые примеси в определенном диапазоне, должны соблюдаться валидированные условия технологических процессов при росте клеток, экспрессии белка и очистке. Для обеспечения отсутствия вирусной контаминации в определенных типах клеток, используемых в производстве, могут потребоваться дополнительные меры. Для лекарственных препаратов, производство которых предусматривает многократные сборы клеток при культивировании, его продолжительность должна находиться в утвержденных пределах.
2. Процессы очистки от нежелательных белков хозяина- продуцента, нуклеиновых кислот, углеводов, вирусов и других примесей должны проводиться в рамках определенных валидированных пределов.
B6. Лекарственные препараты моноклональных антител
1. Моноклональные антитела могут быть произведены из мышиных или человеческих гибридом или с помощью технологий рекомбинантной ДНК. Для обеспечения безопасности и качества лекарственного препарата должны проводиться соответствующие мероприятия по контролю в отношении исходных клеток (в том числе питающих клеток, в случае их использования) и исходных материалов, используемых для создания гибридомы (линии клеток). Следует удостовериться, что данные мероприятия проводятся в утвержденных пределах. Особое внимание следует уделять доказательству отсутствия вирусов в лекарственном препарате. Для доказательства пригодности лекарственных препаратов, произведенных на одной и той же технологической основе, возможно использование данных, полученных при испытании одного из них.
2. Должна быть проведена проверка того, что критерии на промежуточных и конечной стадии производственного процесса контролируются и находятся в утвержденных пределах.
3. Производственные условия для приготовления субфрагментов антител (например, Fab, F(ab’) 2, scFv) и любых других модификаций (например, для введения радиоактивных меток, конъюгации, химического связывания) должны соответствовать валидированным параметрам.
B7. Лекарственные препараты трансгенных животных
Обеспечение постоянства исходного материала, полученного из трансгенного источника, является более проблематичным, чем при использовании стандартных нетрансгенных биотехнологических источников. Следовательно, должны соблюдаться повышенные требования для доказательства постоянства всех свойств лекарственного препарата от серии к серии.
1. Для производства биологических лекарственных препаратов могут использоваться различные виды животных, в том числе могут проводиться забор и очистка биологических жидкостей (например, молока). Животные должны иметь четкую и уникальную маркировку. Должны быть предусмотрены дублирующие меры на случай утраты первичного идентифицирующего маркера.
2. Условия содержания и ухода за животными должны обеспечивать наименьший возможный контакт животных с патогенными агентами и зоонозами. Должны быть разработаны соответствующие меры защиты окружающей среды. Должна быть разработана программа наблюдения за здоровьем животных с соответствующим внесением записей в документацию. Также должны быть расследованы любые инциденты и определено их влияние на возможность дальнейшего использования животного и ранее полученных серий продукции. Следует удостовериться, что любые лекарственные препараты, применявшиеся для лечения животных, не приведут к контаминации производимого лекарственного препарата.
3. Должна существовать документация с родословной от животного- основателя до животных, использующихся для производства. Запрещается смешивание материалов, полученных из разных трансгенных линий животных, так как они происходят от разных животных-основателей.
4. Условия, при которых производится забор материалов, должны соответствовать регистрационному досье и протоколу клинических исследований. График забора материала и условия, при которых животные могут быть исключены из процесса производства лекарственного препарата, должны соответствовать утвержденным процедурам и критериям приемлемости.
B8. Лекарственные препараты трансгенных растений
Обеспечение постоянства исходного материала, полученного из трансгенного источника, является более проблематичным, чем при использовании стандартных нетрансгенных биотехнологических источников. Следовательно, должны соблюдаться повышенные требования для доказательства постоянства всех свойств лекарственного препарата от серии к серии.
1. Для предотвращения контаминации главных и рабочих трансгенных банков посторонними материалами растительного происхождения и соответствующими посторонними агентами могут понадобиться дополнительные меры, предшествующие мероприятиями или следующие за мероприятиями, указанными в части А настоящего приложения. Должен проводиться контроль стабильности гена на протяжении определенного количества генераций.
2. Для обеспечения постоянства сбора урожая от разных культур растений, такие растения должны иметь четкую и уникальную маркировку, и должны быть указаны их основные характеристики. В частности, состояние здоровья растений, входящих в культуру, должно контролироваться с определенной периодичностью на протяжении периода их выращивания.
3. Должны быть установлены меры предосторожности для защиты культур. По возможности следует минимизировать их контаминацию микробиологическими агентами и перекрестную контаминацию растениями другого вида. Должны быть приняты меры для предотвращения контаминации лекарственного препарата такими материалами, как пестициды и удобрения. Должна быть разработана программа мониторинга с соответствующим внесением записей в документацию, также должны быть расследованы любые инциденты и определено их влияние на возможность дальнейшего использования культуры в производственном процессе.
4. Должны быть четко определены условия, определяющие случаи, при которых растения могут быть исключены из производственного процесса. Следует установить пределы приемлемости для материалов (например, основных белков), которые могут помешать процедуре очистки продукции. Должно быть подтверждено, что результаты находятся в пределах утвержденных норм.
5. Должны быть документально оформлены условия окружающей среды (температура, дождь), которые могут повлиять на качественные характеристики лекарственного препарата, а также на производственный выход рекомбинантного белка (от времени посева, на протяжении культивирования и до момента сбора и промежуточного хранения собранных материалов). При оформлении этой документации должны учитываться принципы, указанные в Правилах надлежащего выращивания и сбора растений в соответствии с законодательством.
B9. Лекарственные препараты генной терапии
Существует несколько типов лекарственных препаратов генной терапии (лекарственные препараты генной терапии, содержащие последовательность рекомбинантных нуклеиновых кислот или генетически модифицированный микроорганизм или вирус, и лекарственные препараты генной терапии, содержащие генетически модифицированные клетки), которые охватываются настоящим разделом. В отношении лекарственных препаратов генной терапии на основе клеток могут применяться некоторые положения, изложенные в разделе В10 части В настоящего приложения.
1. Вследствие того, что клетки, используемые для производства лекарственных препаратов генной терапии, получены от людей (аутологичные или аллогенные) или животных (ксеногенные), существует повышенный риск их контаминации побочными агентами. Для изоляции аутологичных материалов, полученных от инфицированных доноров, должны, быть предусмотрены особые мероприятия. Для таких исходных материалов, а также для криопротекторов, питательных сред, клеток и векторов надежность контрольных и испытательных мероприятий должна быть основана на принципах управления рисками для качества и должна соответствовать регистрационному досье. Созданные клеточные линии для производства вирусных векторов и проведения контрольных и испытательных мероприятий должны также основываться на принципах управления рисками для качества. При необходимости, должны использоваться вирусные посевные культуры и системы банков клеток.
2. На возможное содержание примесей, посторонних агентов и перекрестную контаминацию влияют такие факторы, как природа генетического материала, тип вектора (вирусный или невирусный) и тип клеток, что должно быть учтено при разработке общей стратегии минимизации риска. На основе указанной стратегии должен быть разработан технологический процесс, спроектированы производственные и складские помещения и оборудование, разработаны процедуры уборки и деконтаминации, а также упаковки, маркировки и реализации.
3. Производство и испытание лекарственных препаратов генной терапии требуют решения специфических вопросов безопасности и качества готового лекарственного препарата и вопросов безопасности пациентов и персонала. Должен применяться подход, основанный на управлении рисками, для обеспечения безопасности персонала, окружающей среды и пациентов, а также должны быть приняты меры контроля в соответствии с установленным статусом биологической опасности. Меры по обеспечению безопасности должны соответствовать требованиям законодательства государств-членов и, при необходимости, требованиям международного права.
4. Передвижение персонала (включая персонал, занятый контролем качества, и обслуживающий персонал) и потоки материалов, включая те, которые хранят и испытывают (например, исходные материалы, образцы для внутрипроизводственного контроля, образцы готового лекарственного препарата и пробы производственной среды) должны быть организованы на основе принципов управления рисками для качества. По возможности при этом используют однонаправленные потоки. Должно учитываться перемещение между зонами, содержащими различные генетически модифицированные организмы, и зонами, содержащими генетически немодифицированные организмы.
5. При проектировании помещений и оборудования должны быть учтены все возможные специальные процедуры, требующиеся для деконтаминации или очистки от организмов, которые используются при производстве лекарственного препарата. По возможности, программа мониторинга состояния производственной среды должна быть дополнена методами определения присутствия специфических микроорганизмов, культивирование которых производилось.
6. При использовании вирусных векторов с ограниченной способностью к репликации должны приниматься меры по предотвращению попадания вирусов дикого типа, которое может привести к возникновению рекомбинантных векторов, способных к репликации.
7. Должен быть предусмотрен план аварийных мероприятий на случай непредвиденного выброса живых микроорганизмов. Указанный план должен включать в себя описание методов и процедур по изоляции микроорганизмов, защите операторов, уборке, проведению деконтаминации и безопасному возобновлению эксплуатации. Должно быть оценено влияние выброса на лекарственные препараты, находящиеся в непосредственной близости, и на любые другие лекарственные препараты, находящиеся в зонах, подверженных такому выбросу.
8. Должны быть предусмотрены меры для отделения помещений для производства вирусных векторов от других зон. Эффективность мер, используемых для разделения, должна быть доказана. Везде, при необходимости, следует использовать закрытые системы. Должен быть предотвращен выброс вирусного материала при отборе образцов, введении добавок и передаче материалов.
9. Не допускается сопутствующее производство различных векторов генной терапии в одной и той же зоне. Одновременное производство невирусных векторов на одном участке должно контролироваться согласно принципам управления рисками для качества. Должна быть показана эффективность процедур перехода между кампаниями.
10. Для обеспечения прослеживаемости лекарственного препарата от начальных стадий (плазмиды, целевые гены и регуляторные последовательности, банки клеток, а также запас вирусных или невирусных векторов) и до готового лекарственного препарата должно быть в наличии детальное описание производства векторов и генетически модифицированных клеток.
11. Перевозка лекарственных препаратов, содержащих или состоящих из генетически модифицированных организмов, должна осуществляться в соответствии с требованиями законодательства.
12. К переносу генов в клетки-реципиенты, который проводится вне организма, предъявляются следующие требования:
а) процесс переноса должен происходить в помещениях, предназначенных для данных действий и имеющих соответствующий уровень изоляции;
b) необходимо принять меры (включая требования пункта 10 части А настоящего приложения) для уменьшения возможности перекрестной контаминации и перепутывания клеток, полученных от разных пациентов. Должно быть также предусмотрено использование валидированных процедур очистки. Одновременное использование разных вирусных векторов должно контролироваться в соответствии с принципами управления рисками для качества. Не разрешается использование некоторых вирусных векторов (например, ретро- и лентивирусов) для производства генетически модифицированных клеток до доказательства отсутствия в них постороннего вектора, способного к репликации;
c) требования прослеживаемости должны быть соблюдены. Необходимо четкое определение серии продукции, начиная от клеточного сырья и заканчивая контейнером с готовым лекарственным препаратом;
d) физико-химические свойства лекарственных препаратов, при производстве которых используются небиологические средства доставки гена, должны быть исследованы и документально подтверждены.
B10. Лекарственные препараты терапии соматическими клетками и
лекарственные препараты тканевой инженерии
Для лекарственных препаратов генетически модифицированных клеток, которые не определены как лекарственные препараты генной терапии, могут также применяться некоторые положения раздела В9 настоящего приложения.
1. Для производства лекарственных препаратов по возможности должны быть использованы разрешенные источники (например, разрешенные лекарственные средства или медицинские изделия с маркировкой СЕ, обозначающей соответствие европейским стандартам) дополнительных материалов (в частности, клеточных продуктов, биомолекул, биоматериалов, поддерживающих систем, матриц).
2. В случаях, когда изделия (в том числе изделия, производимые на заказ) являются составной частью готовой продукции, должны соблюдаться следующие требования:
a) должно быть принято в форме письменного документа соглашение между производителем лекарственного препарата и производителем медицинского изделия, в котором должна содержаться достаточная информация о медицинском изделии, чтобы избежать изменения его свойств во время производства высокотехнологичного лекарственного средства (ATMP). Указанное соглашение должно содержать требование о контроле изменений, предложенных для медицинского изделия;
b) указанное соглашение должно предусматривать обмен информацией об отклонениях, имевших место при производстве медицинского изделия.
3. Вследствие того, что соматические клетки получены от людей (аутологичные или аллогенные) или животных (ксеногенные), существует повышенный риск их контаминации побочными агентами. Для изоляции аутологичных материалов, полученных от инфицированных доноров, должны быть предусмотрены особые мероприятия. Должна быть обеспечена надежность контрольных и испытательных мероприятий для таких исходных материалов.
4. При невозможности проведения стерилизации готового лекарственного препарата при помощи стандартных методов (например, фильтрацией), стадии технологического процесса должны быть проведены в асептических условиях.
5. Должное внимание следует уделять специфическим требованиям для всех стадий криоконсервации (например, скорости изменения температур во время замораживания и размораживания). Тип камеры хранения, способы размещения материалов и процессы извлечения должны сводить к минимуму риск перекрестной контаминации, обеспечивать качество лекарственных препаратов и способствовать их правильному извлечению. Для обеспечения безопасности работы с лекарственными препаратами, в состав которых входят позитивные серологические маркеры, а также для хранения этих лекарственных препаратов должны использоваться процедуры, принятые в форме письменного документа.
6. Должны быть проведены испытания стерильности на предмет отсутствия бактериальной или грибковой контаминации в культурах клеток или банках клеток, не содержащих антибиотики. Должна также учитываться необходимость определения специфических микроорганизмов, требующих специальных условий культивирования.
7. В случае необходимости должна проводиться программа мониторинга стабильности, включающая в себя наличие контрольных и архивных образцов в количестве, достаточном для проведения дальнейших испытаний.
Определения
В настоящий раздел включены только термины, используемые в настоящем приложении и требующие расширенного объяснения. Приводятся также ссылки на определения, существующие в законодательстве или других источниках. В дополнение к определениям, указанным в настоящем разделе, используется общий словарь терминов и определений Правил надлежащей производственной практики Евразийского экономического союза.
«адъювант» (adjuvant) - химическое или биологическое вещество, усиливающее иммунную реакцию на антиген;
«активная фармацевтическая субстанция» (active substance) - вещество, которое может иметь человеческое (например, донорская кровь или препараты донорской крови), животное (например, микроорганизмы, целые животные, части органов, секреты животных, токсины, экстракты, препараты крови), растительное (например, микроорганизмы, растения, части растений, выделения растений, экстракты), химическое (например, простые вещества, природные соединения, соединения, полученные путем химической модификации или химического синтеза) происхождение;
«аллергены» (allergens) - вещества антигенной или гаптеновой природы, способные сенсибилизировать организм и вызывать аллергию;
«аллергоиды» (allergoids) - химически измененные аллергены с пониженной реактивностью иммуноглобулина Е (IgE);
«антигены» (аlltigens) - вещества (например, токсины, чужеродные белки, бактерии, клетки ткани), способные вызвать специфические иммунные реакции;
«антитело» (аntibody) - белки, произведенные B-лимфоцитами, которые специфически связываются с определенными антигенами. На основе ключевых различий в методах их производства выделяют два главных типа антител: моноклональные и поликлональные антитела;
«банк клеток» (cell bank) - совокупность соответствующих контейнеров, хранящихся в определенных условиях, содержимое которых имеет однородный состав. Каждый контейнер содержит аликвоту одного пула клеток;
«биологическая активная фармацевтическая субстанция» (biological active substance) - активная фармацевтическая субстанция которая произведена с использованием биологического источника или экстрагирована из биологического источника, которая должна быть охарактеризована с использованием физических, химических и биологических испытаний и качество которой определяется этими испытаниями в сочетании с контролем процессов ее производства;
«биологический лекарственный препарат» (biological medicinal product) - лекарственный препарат, активная фармацевтическая субстанция которого является биологической активной фармацевтической субстанцией;
«бионагрузка» (bio burden) - уровень и вид микроорганизмов (неприемлемые или допустимые микроорганизмы), которые содержатся в исходном сырье, питательной среде, биологических активных фармацевтических субстанциях, промежуточных продуктах или готовых лекарственных препаратах;
«вектор» (vector) - агент трансмиссии, переносящий генетическую информацию от одной клетки или организма к другой, например плазмиды, липосомы, вирусы;
«вирусный вектор» (viral vector) - вектор, произведенный путем модификации вируса с помощью методов молекулярной биологии для удерживания некоторых, но не всех, материнских генов вируса. При удалении генов, ответственных за способность вируса к репликации, созданный вектор является неспособным к репликации;
«вне живого организма» (ex-vivo) - процесс, при котором процедуры проводятся на тканях или клетках вне живого организма с последующим возвратом тканей или клеток в живой организм;
«внутри живого организма» (in-vivo) - процедуры, проводимые на живых организмах;
«вспомогательное вещество» (excipient) - вещество, за исключением активных фармацевтических субстанций, входящее в состав лекарственного препарата для придания ему необходимых свойств;
«гаптен» (hapten) - молекула с низкой молекулярной массой, не являющаяся антигеном по своей природе до момента конъюгации с «молекулой-носителем»;
«ген» (gene) - последовательность ДНК, кодирующая один или несколько белков;
«генетически модифицированный организм» (ГМО) (genetically modified organism (GMO)) - любой организм, кроме человека, с измененным генетическим материалом, отличающимся от генетического материала, который получается при естественном спаривании и (или) естественной рекомбинации;
«гибридома» (hybridoma) - иммортализованная («бессмертная») линия клеток, производящая желаемые (моноклональные) антитела и обычно получаемая путем искусственного слияния В-лимфоцитов с опухолевыми клетками;
«главный банк клеток» (master cell bank, (MCB)) - аликвота одного пула клеток, которая, как правило, была получена из конкретного клеточного клона при определенных условиях, распределена во множество контейнеров и хранится при определенных условиях;
«главный вирусныйпосевной материал» (master virus seed, (MVS)) - аликвота индивидуального субштама вирусов, которая, как правило, была получена из конкретного вирусного штама при определенных условиях, распределена во множество контейнеров и хранится при определенных условиях;
«главный трансгенный банк» (master transgenic bank) - аналогично определению главного банка клеток, но в отношении трансгенных растений или животных;
«закрытая система» (closed system) - система, в которой активная фармацевтическая субстанция или лекарственный препарат не подвержены непосредственному влиянию производственной среды;
«зона» (area) - определенный набор помещений в пределах одного здания, в которых производится один или несколько лекарственных препаратов и которые имеют отдельную систему воздухоподготовки;
«зооноз» (zoonosis) - инфекционные и инвазионные заболевания животных, которые при определенных условиях могут передаваться человеку;
«использование в условиях изоляции» (contained use) - любая деятельность, при которой генетически модифицированные организмы получают, культивируют, хранят, транспортируют, разрушают, уничтожают или используют каким-либо способом и при которой используются специальные меры изоляции для ограничения распространения этих организмов и обеспечения безопасности населения и окружающей среды;
«исходное сырье» (raw materials) - любые субстанции, используемые для производства или экстрагирования активных фармацевтических субстанций, но не являющиеся их источником, такие как реагенты, среды для культивирования, сыворотка телячьих эмбрионов, добавки и буферы для хроматографии и др.;
«исходные материалы» (starting materials) - все материалы, из которых активная фармацевтическая субстанция производится или экстрагируется. Для биологических лекарственных препаратов исходные материалы представляют собой любые субстанции биологического происхождения, такие как микроорганизмы, органы и ткани растительного или животного происхождения, клетки или жидкости (включая кровь и плазму) человеческого или животного происхождения, а также биотехнологические клеточные субстраты (рекомбинантные и природные), включая первичные клетки;
«клеточный запас» (сell stock) - первичные клетки, размножившиеся до заданного количества клеток, аликвоты которых отбираются и используются в качестве исходного материала для производства ограниченного количества серий лекарственных препаратов на основе клеток;
«моноклональные антитела» (monoclonal antibodies (MAb)) - гомогенная популяция антител, способных присоединяться к единственному эпитопу (антигенной детерминанте), полученная из единственного клона лимфоцитов или с помощью технологии рекомбинантной ДНК;
«ответственное лицо» (Responsible Person, (RP)) - данное определение используется в значении, установленном приложением № 14 к Правилам надлежащей производственной практики Евразийского экономического союза;
«перенос генов (gene transfer)» - процесс переноса гена в клетки, включающий в себя систему экспрессии, содержащуюся в системе доставки, которая называется вектором. Вектор может быть как вирусного, так и невирусного происхождения. После переноса генов, генетически модифицированные клетки также могут иметь название «трансформированные клетки»;
«питающие клетки» (feeder cells) - клетки, используемые в комбинированной культуре для поддержания плюрипотентности (способности дифференцироваться в множество специализированных типов клеток) стволовых клеток. Для культуры человеческих эмбриональных стволовых клеток типичные питающие слои состоят из эмбриональных фибробластов мыши (ЭФМ) или эмбриональных фибробластов человека (ЭФЧ), в которых специальными методами предотвращено деление;
«плазмида» (plasmid) - часть ДНК, обычно существующая в бактериальной клетке в виде кольцевой структуры, отделенной от клеточной хромосомы. Плазмида может быть модифицирована с помощью методов молекулярной биологии, выделена из бактериальной клетки и использована для переноса и встраивания ее ДНК в геном другой клетки;
«поддерживающая система» (scaffold) - средство поддержки, средство доставки или матрица, которые обеспечивают структуру или содействуют миграции, связыванию или транспорту клеток и (или) биологически активных молекул;
«поликлональные антитела» clonal antibodies) - антитела, полученные от нескольких клонов лимфоцитов и выработанные человеческим или животным организмом в ответ на введение антигена;
«помещение для производства нескольких лекарственных препаратов» (multi-product facility) - помещение для последовательного производства или производства по принципу кампаний различных групп биологических активных фармацевтических субстанций и лекарственных препаратов и в котором комплект используемого оборудования может быть специализированным или неспециализированным для каждого отдельного типа субстанций или лекарственных препаратов;
«преднамеренный выброс» (deliberate release) - преднамеренный выпуск в окружающую среду генетически модифицированных организмов, для которых не используются специальные меры изоляции для ограничения распространения этих организмов и обеспечения безопасности населения и окружающей среды;
«производство по принципу кампаний» (campaigned manufacture) - последовательное производство ряда серий одного и того же лекарственного препарата в течение определенного периода времени, после которого проводятся строгие контрольные мероприятия перед переключением на производство другого лекарственного препарата. Лекарственные препараты не производятся одновременно, но для их производства может быть использовано одно и то же оборудование;
«промежуточная продукция» (intermediate product) - данное определение используется в значении, установленном разделом «Термины и определения» Правил надлежащей производственной практики Евразийского экономического союза и разделом «Термины и определения» части II Правил надлежащей производственной практики Евразийского экономического союза;
«процедура ретроспективного анализа» (look-back) - документально оформленная процедура, обеспечивающая отслеживание биологических активных фармацевтических субстанций или лекарственных препаратов несоответствующего качества вследствие использования выбракованных животных или человеческих материалов из-за присутствия в этих материалах контаминирующего агента или при выявлении негативных факторов у животных или людей, являющихся источником этих материалов;
«рабочий банк клеток» (working cell bank(WCB)) - гомогенный пул микроорганизмов или клеток, полученных из главного банка клеток и однородно распределенных в определенное число контейнеров. Рабочий банк клеток хранится в условиях, обеспечивающих его стабильность и использование в производстве;
«рабочий вирусный посевной материал» (working virus seed (WVS) ) - гомогенный пул вирусов, полученных из главного вирусного посевного материала и однородно распределенных в определенное число контейнеров. Рабочий вирусный посевной материал хранится в условиях, обеспечивающих его стабильность и использование в производстве;
«рабочий трансгенный банк» (working transgenic bank) - гомогенный пул клеток трансгенных растений или животных, полученных из главного трансгенного банка и однородно распределенных в определенное число контейнеров. Рабочий трансгенный банк хранится в условиях, обеспечивающих его стабильность и использование в производстве;
«свободные от специфических патогенов» (specified pathogen free (SPF)) - животные материалы (например, куры, эмбрионы или культуры клеток), использующиеся для производства или контроля качества биологических лекарственных препаратов, полученные из групп животных (например, стад или стай), свободных от определенных патогенов. Такие стада или стаи определяются как группы животных, которые живут в общей среде и имеют ухаживающий за ними персонал, который не пребывает в контакте с животными, не свободными от специфических патогенов;
«соматические клетки» (somatic cells) - все клетки тела человека или животного, кроме репродуктивных клеток. Эти клетки могут быть аутологичными (от того же пациента), аллогенными (от другого человека) или ксеногенными (от животного) соматическими живыми клетками, манипуляции с которыми или изменение которых проводились в условиях вне живого организма с последующим введением в организм человека для достижения терапевтического, диагностического или профилактического действия;
«трансгенный» (transgenic) - организм, содержащий в своей обычной генетической структуре чужеродный ген для экспрессии биологических фармацевтических материалов;
«уровень биологической безопасности» (biosafety level (BSL)) - условия изоляции, требующиеся для безопасной работы с микроорганизмами разных уровней патогенности, начиная от 4-й группы патогенности (наименьший риск, маловероятно приводящий к заболеванию человека) и до 1-й группы патогенности (наивысший риск, вызывающий тяжелые легко распространяемые заболевания);
«чистая культура» (аксеничная культура) (mono sepsis (axenic)) - культура, содержащая одинаковые микроорганизмы и не контаминированная любыми другими организмами.
ПРИЛОЖЕНИЕ № 3
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству радиофармацевтических
лекарственных препаратов
Принцип
Производство радиофармацевтических лекарственных средств должно быть организовано в соответствии с принципами, приведенными в частях I и II Правил надлежащей производственной практики Евразийского экономического союза. Настоящее приложение устанавливает правила, специфические для производства радиофармацевтических лекарственных средств.
Настоящее приложение не распространятся на изготовление радиофармацевтических лекарственных средств в специализированных аптеках (в больничных или действующих самостоятельно) с использованием радионуклидных генераторов и наборов реагентов в соответствии с законодательством государств - членов Евразийского экономического союза (далее - государства-члены).
В соответствии с требованиями радиационной безопасности ответственность за применение радиации в медицинских целях лежит на медицинских работниках. При применении радиофармацевтических лекарственных средств в диагностических и терапевтических целях должно быть обеспечено, при необходимости, наличие специалиста по медицинской физике.Настоящее приложение распространяется также на радиофармацевтические лекарственные средства, использующиеся в клинических исследованиях.
Транспортирование радиофармацевтических лекарственных средств выполняется в соответствии с требованиями по радиационной безопасности Международного агентства по атомной энергии (МАГАТЭ) и требованиями законодательства государств-членов.
Могут использоваться методы, отличающиеся от приведенных в настоящем приложении, но позволяющие выполнять требования по обеспечению качества продукции. Иные методы должны быть валидированы и обеспечивать уровень качества, по крайней мере, эквивалентный требованиям настоящего приложения.
Введение
1. Производство радиофармацевтических лекарственных средств и обращение с ними представляет потенциальную опасность. Уровень рисков зависит, в частности, от типа ионизирующего излучения, энергии излучения и периода полураспада радионуклидов. Особое внимание следует уделять предотвращению перекрестной контаминации, хранению остатков радиоактивных материалов и удалению отходов.
2. Из-за того, что некоторые радионуклиды имеют короткий срок хранения, допускается выпускать в обращение содержащие их радиофармацевтические лекарственные средства до завершения испытаний контроля качества. В этом случае в специальной процедуре должен быть четко и подробно определен порядок выдачи разрешения на выпуск, включая ответственность персонала и непрерывную оценку эффективности системы обеспечения качества.
3. Областью применения настоящего приложения является деятельность промышленных производств, ядерных центров, институтов и ПЭТ-центров по производству и контролю качества следующих типов продукции:
радиофармацевтических лекарственных средств;
радиофармацевтических лекарственных средств для ПЭТ (позитронно-эмиссионной томографии);
радиоактивных предшественников для производства радиофармацевтических лекарственных препаратов;
радионуклидных генераторов.
|
Вид производства |
Действие не распространяется* |
Следует выполнять требования частей II и I Правил надлежащей производственной практики Евразийского экономического союза (по мере приближения стадии производства к готовому продукту требования усиливаются), включая соответствующие приложения |
|||
|
Радиофармацевти ческие лекарственные средства; радиофармацевти ческие лекарственные средства для ПЭТ; радиоактивные предшественники |
продукция реакторов и** циклотронов |
химический синтез |
стадии очистки |
обработка, приготовление, дозирование |
асептическое производство или финишная стерилизация |
|
Радионуклидные генераторы |
продукция реакторов и циклотронов2 |
технологический процесс (сборка колонки, генератора, зарядка генератора и др.) |
|||
*Мишень и система передачи от циклотрона к установке синтеза могут рассматриваться как первая стадия производства активных фармацевтических субстанций.
**Продукция, полученная в результате радиохимического выделения радионуклида из облученной радиоактивной мишени.
***Продукция, полученная в результате радиохимического выделения материнского радионуклида из облученной радиоактивной мишени.
4. Производитель готового радиофармацевтического лекарственного средства должен иметь описание технологического процесса производства активной фармацевтической субстанции, готового радиофармацевтического лекарственного средства и указать, какие требования Правил надлежащей производственной практики Евразийского экономического союза (часть I или II) распространяются на различные технологические операции (стадии).
5. Производство радиофармацевтических лекарственных средств должно выполняться в соответствии с требованиями норм радиационной безопасности.
6. Производство радиофармацевтических лекарственных средств, предназначенных для парентерального введения, должно выполняться в соответствии с требованиями, предъявляемыми к стерильности таких лекарственных средств, с соблюдением в соответствующих случаях асептических условий производства в соответствии с приложением № 1 к Правилам надлежащей производственной практики Евразийского экономического союза.
7. Спецификации и методы контроля качества радиофармацевтических лекарственных средств устанавливаются в Фармакопее Евразийского экономического союза либо в национальных фармакопеях государств- членов или в регистрационных досье на эти лекарственные средства.
Клинические исследования
8. На производство радиофармацевтических лекарственных средств, предназначенных для клинических исследований, распространяются также требования приложения № 13 к Правилам надлежащей производственной практики Евразийского экономического союза.
Обеспечение качества
9. Обеспечение качества при производстве радиофармацевтических лекарственных средств имеет особое значение в виду их специфических особенностей, малых объемов серий и в некоторых случаях необходимости их медицинского применения до завершения контроля качества.
10. Защита продукции от контаминации и перекрестной контаминации должна быть обеспечена так же, как и при производстве любых лекарственных средств. Но в данном случае предъявляется дополнительное требование по защите производственной среды и персонала от ионизирующего излучения. Это означает, что система обеспечения качества приобретает исключительно важное значение.
11. Очень важным является тщательная регистрация данных мониторинга помещений и процессов. Оценка этих данных является частью процесса выпуска серии в обращение.
12. При производстве радиофармацевтических лекарственных средств следует в необходимом объеме проводить квалификацию и валидацию. Для определения объема работ по квалификации и валидации должен применяться подход, основанный на управлении рисками, с особым вниманием к комбинации требований Правил надлежащей производственной практики Евразийского экономического союза и радиационной безопасности.
Персонал
13. Все технологические операции должны выполняться персоналом, имеющим специальную подготовку по радиационной безопасности. Персонал, занятый в производстве, контроле качества и выпуске радиофармацевтических лекарственных средств, должен пройти соответствующее обучение, связанное с особенностями системы обеспечения качества радиофармацевтических лекарственных средств. Уполномоченное лицо несет полную ответственность за выпуск радиофармацевтических лекарственных средств.
14. Персонал, работающий в зонах производства радиофармацевтических лекарственных средств (включая занятый уборкой и техническим обслуживанием), должен пройти дополнительное обучение, связанное со спецификой процессов и продукции.
15. Если производственные помещения и оборудование используются также для проведения исследований, то исследовательский персонал должен пройти обучение Правилам надлежащей производственной практики Евразийского экономического союза. Служба обеспечения качества должна рассматривать и давать разрешение на проведение работ, связанных с исследованиями, для того чтобы исключить их опасное влияние на производство.
Помещения и оборудование
Общие положения
16. Производство радиофармацевтических лекарственных средств должно проводиться в контролируемых зонах, в которых выполняются требования к производственной среде и радиационной безопасности. Все технологические операции должны выполняться в помещениях и на оборудовании, специально предназначенных для производства радиофармацевтических лекарственных средств.
17. Следует принять меры по предотвращению перекрестной контаминации от персонала, исходного сырья и материалов, радионуклидов и пр. Везде, где это возможно, следует использовать закрытое или изолированное оборудование. При использовании оборудования открытого типа или при открывании оборудования следует принять меры по сведению риска контаминации к минимуму. При оценке рисков следует показать, что чистота производственной среды удовлетворяет требованиям, предъявляемым к типу выпускаемой продукции.
18. Вход в производственные зоны должен осуществляться через помещения для переодевания (санпропускники) и должен быть разрешен исключительно для персонала, имеющего право доступа в них.
19. Следует проводить мониторинг рабочих мест и производственной среды в отношении уровня радиации, концентрации частиц и микроорганизмов. Порядок проведения мониторинга устанавливается при квалификации эксплуатации (PQ).
20. Для гарантии того, что используемые помещения и оборудование являются соответствующими и прошедшими квалификацию, необходимо проводить профилактическое техническое обслуживание, калибровку и квалификацию. Эти работы должны выполняться подготовленным персоналом, а факт их проведения и полученные результаты должны оформляться в форме письменного документа.
21. Следует принять меры по защите производственной зоны от радиоактивного загрязнения. Следует организовать надлежащий контроль радиоактивных загрязнений прямым методом с помощью дозиметров или косвенно - методом смывов в установленном порядке.
22. Поверхности оборудования, соприкасающиеся с продуктом, не должны вступать с ним в реакцию, не должны ничего выделять и не должны абсорбировать продукт, чтобы таким образом не изменить качество радиофармацевтического лекарственного средства.
23. Рециркуляция воздуха из помещений, в которых выполняется работа с радиофармацевтическими лекарственными средствами, не допускается, за исключением случаев, когда применение рециркуляции обосновано. В вытяжных системах должна быть предусмотрена защита от загрязнения окружающей среды радиоактивными частицами и газами. В контролируемых зонах должна быть предусмотрена защита от контаминации частицами и микроорганизмами.
24. В целях недопущения распространения радиоактивных частиц, в зонах, где находится открытый продукт, необходимо поддерживать отрицательное давление по отношению к окружающим зонам. В то же время следует защитить продукт от контаминации из производственной среды. Это может быть достигнуто за счет применения барьерной технологии и воздушных шлюзов, работающих по принципу каскада давлений.
Производство стерильной продукции
25. Стерильные радиофармацевтические лекарственные средства разделяются на две группы: лекарственные препараты, выпускаемые в асептических условиях, и лекарственные препараты, подлежащие финишной стерилизации. В производстве должен поддерживаться уровень чистоты производственной среды, соответствующий виду выполняемых операций. Должны соблюдаться требования к чистоте рабочих зон, в которых продукция или первичная (внутренняя) упаковка может находиться в контакте с окружающим воздухом, приведенные в приложении № 1 к Правилам надлежащей производственной практики Евразийского экономического союза.
26. Для определения требований к перепадам давления, направлению потока воздуха и его качеству могут использоваться методы оценки рисков.
27. В закрытых автоматизированных системах, представляющих собой, как правило, горячие камеры с размещением в них установок химического синтеза, систем очистки, стерилизующей фильтрации «на линии», достаточно обеспечить класс чистоты C. В горячие камеры, находящиеся в закрытом состоянии, должен подаваться воздух после фильтрации с высокой степенью чистоты. Асептические операции должны выполняться в помещении (зоне) класса A.
28. До начала производства сборка стерильного оборудования и компонентов (трубок, стерилизующих фильтров), подсоединение линий подачи жидкостей к укупоренным герметичным стерильным флаконам, должны выполняться в асептических условиях.
Документация
29. Все документы, касающиеся производства радиофармацевтических лекарственных средств, должны быть разработаны, согласованы, утверждены и выданы в соответствии с процедурой, принятой в форме письменного документа.
30. Требования к исходному сырью, упаковочным материалам, материалам для маркировки, критическим для качества промежуточным материалам и готовым радиофармацевтическим лекарственным средствам должны быть указаны в спецификациях. Должны быть также спецификации на критические для качества материалы и компоненты (вспомогательные материалы, уплотнения, наборы для стерилизующей фильтрации и др.), используемые в процессе производства и способные оказать критическое влияние на качество продукции.
31. Для радиофармацевтических лекарственных средств следует установить критерии приемлемости, включая спецификации на момент выпуска и на период срока годности (например, для радиохимической чистоты, объемной активности, радионуклидной чистоты и удельной активности).
32. В записях по использованию, очистке, дезинфекции или стерилизации, техническому обслуживанию основного оборудования следует указывать дату и время выполнения операции, должна быть подпись лица, выполнившего работу, и, при необходимости, следует указывать наименование продукции и номер серии.
33. Записи следует хранить не менее 3 лет, если иное не установлено законодательством государств-членов.
Производство
34. С целью сведения к минимуму риска перекрестного загрязнения радиоактивными веществами или перепутывания материалов не допускается одновременное производство различных радиофармацевтических лекарственных средств в одной рабочей зоне (горячей камере, ламинарной зоне или шкафу).
35. Особое внимание следует уделять валидации, включая валидацию компьютеризированных систем в соответствии с приложением № 11 к Правилам надлежащей производственной практики Евразийского экономического союза. Новые процессы должны пройти перспективную валидацию.
36. Критические параметры следует, как правило, определять до или в процессе проведения валидации. При этом следует определять допустимые предельные значения изменений параметров, необходимые для стабильного производства.
37. Для продуктов, наполняемых в асептических условиях, следует проводить контроль целостности мембранных фильтров, с учетом необходимости обеспечения радиационной безопасности и сохранения стерильности фильтров.
38. Учитывая радиационную активность готовой продукции, допускается наносить маркировку на первичную упаковку до начала производства. На стерильные пустые закрытые флаконы может быть нанесена маркировка с частичной информацией до операции наполнения, при этом стерильность не должна быть нарушена и не должно быть помех для визуального контроля наполненных флаконов.
Контроль качества
39. Некоторые радиофармацевтические лекарственные средства могут быть выпущены и использованы на основе оценки документации на серию до завершения всех химических и микробиологических испытаний.
Оформление разрешения на выпуск радиофармацевтических лекарственных средств может быть выполнено в 2 и более этапов до и после завершения аналитического контроля в полном объеме:
a) оценка назначенным лицом записей по производству серии, которое должно охватывать условия производства и аналитический контроль, проведенные до момента разрешения транспортировки радиофармацевтического лекарственного средства в статусе «карантин» в клиническое подразделение;
b) Уполномоченное лицо выдает разрешение на выпуск после проведения оценки окончательных результатов аналитического контроля, всех отклонений от нормального процесса, которые должны быть оформлены документально, обоснованы и утверждены. Если некоторые результаты контроля невозможно получить до использования лекарственного средства, то Уполномоченному лицу следует оформить разрешение на выпуск лекарственного средства условно до начала его использования и окончательно оформить разрешение на выпуск лекарственного средства после получения всех результатов контроля.
40. Большинство радиофармацевтических лекарственных средств используется в течение короткого периода времени, что обусловлено коротким периодом полураспада радионуклидов, поэтому их срок годности должен быть четко указан.
41. Радиофармацевтические лекарственные средства, содержащие радионуклиды с большим периодом полураспада, следует контролировать на соответствие всем требованиям до оформления разрешения на выпуск Уполномоченным лицом.
42. Контроль проб может быть проведен не сразу после их отбора, чтобы обеспечить требуемое снижение уровня активности. Все виды контроля, включая контроль на стерильность, должны быть проведены как можно быстрее.
43. Процедура, принятая в форме письменного документа, должна устанавливать порядок оценки продукции и результатов контроля до отправки продукции.
44. Продукция, не соответствующая установленным требованиям, должна быть отклонена. Если предусмотрена переработка материала, то она должна выполняться по заранее утвержденной процедуре. Готовая продукция должна соответствовать установленным требованиям, что должно быть подтверждено до ее выпуска. Не допускается переработка возвращенной продукции, с которой следует обращаться как с радиоактивными отходами.
45. В специальной процедуре должен быть определен порядок действий Уполномоченного лица в случае обнаружения несоответствия продукции требованиям спецификации после ее отгрузки до истечения срока годности. Такие случаи должны быть расследованы, должны быть выполнены необходимые предупреждающие и корректирующие мероприятия для недопущения подобных ситуаций в будущем. Этот процесс должен быть документально оформлен.
46. При необходимости следует информировать ответственный персонал медицинского учреждения. Для содействия этому должна быть обеспечена прослеживаемость в отношении радиофармацевтических лекарственных средств.
47. Должен быть установлен порядок контроля исходного сырья и материалов. При выборе и утверждении поставщика следует убедиться в том, что поставляемые им исходное сырье и материалы неизменно соответствуют требованиям спецификаций. Исходное сырье, упаковочные материалы и вспомогательные материалы для критических процессов должны приобретаться только у утвержденных поставщиков.
Контрольные и архивные образцы
48. От каждой серии нерасфасованных радиофармацевтических лекарственных средств должно быть отобрано достаточное количество образцов, которые должны храниться не менее 6 месяцев с момента истечения срока годности готовой продукции, если иное не установлено в процессе управления рисками.
49. Образцы используемого в производстве исходного сырья, за исключением растворителей, газов и воды, должны храниться не менее 2 лет после выпуска продукции. Этот срок может быть сокращен, если в спецификации на сырье указан более короткий период стабильности.
50. По согласованию с уполномоченным органом может быть определен иной порядок отбора и хранения проб исходного сырья, материалов, продукции, произведенных по индивидуальному заказу или в малых количествах или если их хранение может вызвать особые трудности.
Реализация
51. Для радиофармацевтических лекарственных средств допускается реализация готовой продукции в контролируемых условиях до получения результатов всех необходимых испытаний. При этом должно быть гарантировано, что лекарственный препарат не будет применен в медицинском учреждении до получения удовлетворительных результатов испытаний и их оценки Уполномоченным лицом, с учетом положений пункта 39 настоящего приложения.
Определения
«горячая камера» (hot-cell) - экранированное рабочее место для производства и обращения с радиоактивными материалами. Горячая камера не обязательно должна быть изолятором;
«приготовление» (preparation) - подготовка набора в медицинском учреждении путем внесения в него радионуклида, элюированного из генератора, или с помощью радиоактивных предшественников. Наборы, генераторы и радиоактивные предшественники должны быть зарегистрированы в установленном порядке;
«радиофармацевтические лекарственные средства» - лекарственные средства, которые содержат в готовой для использования форме один радионуклид или несколько радионуклидов (радиоактивных изотопов);
«радиоактивный предшественник» (radiopharmaceutical precursor) - радиоактивное вещество, предназначенное для введения радионуклидной метки в другое вещество перед его применением;
«уполномоченное лицо» (qualified Person) -лицо, назначенное производителем лекарственных средств, которое осуществляет подтверждение соответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза. Обязанности уполномоченного лица детально описаны в главе 2части I и приложении № 16 к Правилам надлежащей производственной практики Евразийского экономического союза.к Правилам надлежащей производственной практики Евразийского экономического союза.
ПРИЛОЖЕНИЕ № 4
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству ветеринарных лекарственных средств (кроме
иммунобиологических ветеринарных лекарственных средств)
1. При производстве ветеринарных лекарственных средств следует соблюдать требования правил надлежащей производственной практики, утверждаемых Евразийской экономической комиссией (далее - правила).
2. Настоящим приложением устанавливаются специальные требования к производству отдельных групп ветеринарных лекарственных средств (ветеринарных лекарственных препаратов против эктопаразитов, ветеринарных лекарственных средств содержащих пенициллины и ветеринарных лекарственных средств подлежащих финишной стерилизации).
3. Производство остальных групп ветеринарных лекарственных средств осуществляется в соответствии с требованиями, установленными другими приложениями к правилам.
4. При производстве ветеринарных лекарственных средств могут использоваться в качестве исходного сырья фармацевтические субстанции, которые внесены в единый реестр зарегистрированных лекарственных средств для медицинского применения Евразийского экономического союза.
Производство лекарственных препаратов против эктопаразитов
5. Несмотря на ограничение, указанное в пункте 3.6 части I Правил надлежащей производственной практики Евразийского экономического союза, лекарственные препараты против эктопаразитов, предназначенные для наружного применения, относящиеся к ветеринарным лекарственным средствам и включенные в лицензию на производство, могут производиться и наполняться в зонах, предназначенных для производства пестицидов, по принципу разделенных во времени циклов производства. Однако в этих помещениях не должны производиться другие виды ветеринарных лекарственных средств.
6. Для предотвращения перекрестной контаминации следует использовать соответствующие валидированные методы очистки. Следует принять меры по обеспечению безопасного хранения ветеринарных лекарственных препаратов в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза.
Производство ветеринарных лекарственных средств, содержащих
пенициллины
7. Использование пенициллинов в ветеринарии не представляет такого риска с точки зрения гиперсенсибилизации животных, как в случае их использования человеком. Зарегистрированные случаи гиперсенсибилизации у лошадей и собак были обусловлены другими токсичными веществами (например, ионофорными антибиотиками для лошадей). Такие лекарственные препараты рекомендуется производить в специально предназначенных изолированных помещениях (пункт 3.6 части I Правил надлежащей производственной практики Евразийского экономического союза), но эта рекомендация может не распространяться на случаи, когда помещения предназначены только для производства ветеринарных лекарственных средств. Однако необходимо принять все необходимые меры по предотвращению перекрестной контаминации и обеспечению безопасности персонала в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза. При использовании общих помещений производство продукции, содержащей пенициллины, должно быть организовано по принципам разделенных во времени циклов производства и должно сопровождаться соответствующими валидированными методиками деконтаминации и очистки.
Хранение образцов
Требования настоящего раздела применяются в дополнение к требованиям, установленным подпунктом viii пункта 1.4и пунктом 6.14 части 1 Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила).
8. В связи с большими объемами окончательных упаковок некоторых ветеринарных лекарственных средств для производителя может оказаться неудобным хранить образцы каждой серии продукции в их окончательной упаковке. Однако производитель должен обеспечить хранение достаточного количества архивных образцов каждой серии продукции в соответствии с требованиями Правил.
9. В любом случае, упаковка для хранения архивных образцов должна быть произведена из того же материала, что и первичная упаковка, в которой этот продукт реализуется на рынке.
Стерильные ветеринарные лекарственные средства
10. Ветеринарные лекарственные средства, подлежащие финишной стерилизации, могут производиться в чистых зонах более низкого класса, чем это требуется в приложении № 1 к Правилам надлежащей производственной практики Евразийского экономического союза (если решение об этом принято уполномоченным органом), однако класс чистоты помещений должен быть не ниже D.
ПРИЛОЖЕНИЕ № 5
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству иммунобиологических ветеринарных
лекарственных средств
Принцип
Производство иммунобиологических ветеринарных лекарственных средств имеет ряд особенностей, которые следует принимать во внимание при внедрении и оценке эффективности системы обеспечения качества.
Из-за большого количества видов животных и сопутствующих патогенных микроорганизмов спектр продукции, производимой на предприятии, может быть очень широким при небольшом объеме производства. В связи с этим производство обычно организуется по принципу отдельных циклов. Более того, ввиду особенностей этого производства (стадии культивирования, отсутствие финишной стерилизации и др.) необходима тщательная защита продукции от контаминации и перекрестной контаминации. Производственная среда должна быть защищена, особенно при использовании патогенных или экзотических микроорганизмов. При использовании микроорганизмов, патогенных для человека, следует особенно тщательно защищать работающий персонал.
С учетом этих факторов, а также вариабельности свойств иммунобиологических лекарственных препаратов и относительно низкой эффективности проводимых испытаний для получения достаточной информации о продукции (в частности, при контроле качества готовой продукции) система обеспечения качества приобретает особенно важное значение. При производстве иммунобиологических ветеринарных лекарственных средств особую роль играет постоянный контроль за соблюдением всех требований, указанных в части I Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила) и настоящем документе. Следует проводить непрерывную оценку данных, получаемых в ходе контроля различных аспектов производства (оборудование, помещения, продукция и др.) на соответствие требованиям Правил. Решения и действия, предпринятые на основании этой оценки, должны быть оформлены документально.
Персонал
1. Все сотрудники (включая персонал, занимающийся очисткой и обслуживанием), работающие в зонах производства иммунобиологической продукции, должны пройти обучение по личной гигиене и микробиологии, а также дополнительное обучение в соответствии со спецификой производимой продукции.
2. Лица, ответственные за производство и контроль качества, должны иметь базовую подготовку по следующим предметам (всем или некоторым): бактериология, биология, биометрия, химия, иммунология, медицина, паразитология, фармация, фармакология, вирусология и ветеринария, а также необходимые знания по защите окружающей среды.
3. Персонал должен быть защищен от возможного заражения микроорганизмами, используемыми при производстве. При использовании микроорганизмов, являющихся возбудителями болезней человека, необходимо принимать особые меры по защите персонала, работающего с этими микроорганизмами или экспериментальными животными.
В случае необходимости персонал должен пройти вакцинацию, а также должен проходить медицинское обследование.
4. Необходимо принимать соответствующие меры, направленные на предотвращение переноса микроорганизмов людьми за пределы производственной зоны. В зависимости от вида микроорганизмов к таким мерам относятся полное переодевание и обязательное принятие душа перед выходом из производственной зоны.
5. При производстве иммунобиологической продукции особую опасность представляет риск контаминации и перекрестной контаминации, вызываемой персоналом.
Для предотвращения контаминации, вызываемой персоналом, предусматривается выполнение ряда мер и процедур, включающих в себя использование защитной одежды на различных стадиях технологического процесса.
Для предотвращения перекрестной контаминации персонал должен перемещаться из одной зоны в другую с соблюдением правил, исключающих риск контаминации. Эти правила должны быть изложены в инструкции. В течение рабочего времени персонал не должен переходить из зон, где возможна контаминация живыми микроорганизмами или где содержатся животные, в помещения, где ведется работа с другими продуктами или микроорганизмами. Если этого избежать невозможно, персонал, занятый в таком производстве, должен следовать четко установленным методикам деконтаминации, в том числе выполнять смену одежды, обуви, и, по мере необходимости, принимать душ.
Считается, что персонал не сталкивается с риском контаминации при входе в изолированную зону, где культуры микроорганизмов находятся в герметически закрытых контейнерах, поверхность которых прошла деконтаминацию, если в этой зоне в течение последних 12 часов не велись работы с открытыми микроорганизмами (за исключением работы с экзотическими микроорганизмами).
Помещения
6. При проектировании помещений должна быть предусмотрена защита как продукции, так и производственной среды. Это может быть достигнуто за счет использования изолированных, чистых, чистых изолированных или контролируемых зон.
7. Операции с живыми микроорганизмами должны проводиться в изолированных зонах. Уровень изоляции должен зависеть от патогенности микроорганизмов и оттого, были ли они классифицированы как экзотические.
8. Операции с инактивированными микроорганизмами следует проводить в чистых зонах. Чистые зоны следует также использовать при работе с неинфицированными клетками, выделенными из многоклеточных организмов, и в некоторых случаях - при работе со средами, прошедшими стерилизующую фильтрацию.
9. Операции с открытыми продуктами или компонентами первичной упаковки, которые не подлежат дальнейшей стерилизации, следует проводить в боксе (установке) с однонаправленным (ламинарным) потоком воздуха класса A, находящемся в зоне класса B.
10. Если производство иммунобиологических ветеринарных лекарственных средств осуществляется в одном здании, то другие операции с живыми микроорганизмами (контроль качества, исследования и диагностика и др.) должны выполняться в отдельных изолированных помещениях. Степень изоляции зависит от патогенности данного вида микроорганизмов и оттого, был ли он классифицирован как экзотический. При выполнении диагностических операций существует риск внесения высокопатогенных микроорганизмов. Уровень изоляции должен соответствовать всем вышеперечисленным рискам. Изоляция может потребоваться также в случаях, если контроль качества и другие операции проводятся в зданиях, расположенных вблизи производственных зданий.
11. Изолированные помещения должны быть легко дезинфицируемыми и должны выполняться следующие требования:
a) отсутствие прямого выхода вентилируемого воздуха наружу;
b) наличие вентиляции с обеспечением отрицательного перепада давления (разрежения). Воздух должен удаляться через НЕРА-фильтры. Рециркуляция воздуха не допускается, за исключением случаев рециркуляции в той же самой зоне при условии дополнительной очистки воздуха НЕРА-фильтрами (обычно это условие выполняется при прохождении рециркулирующего воздуха через приточные НЕРА- фильтры, которые используются для этой зоны). Поступление воздуха из одной зоны в другую допускается в случае, если вытяжной воздух проходит через два последовательно установленных НЕРА-фильтра. При этом должны быть предусмотрены непрерывный контроль целостности первого фильтра и средства безопасного удаления вытяжного воздуха в случае повреждения этого фильтра;
c) воздух, выходящий из производственных зон, в которых проводится работа с экзотическими микроорганизмами, должен проходить через 2 последовательно установленных НЕРА-фильтра. При этом рециркуляция воздуха между производственными зонами не допускается;
d) наличие системы сбора и дезинфекции жидких отходов, включая контаминированный конденсат из стерилизаторов, биореакторов и др. Твердые отходы, в том числе трупы животных, следует дезинфицировать, стерилизовать или сжигать. Контаминированные фильтры необходимо удалять безопасным способом;
e) комнаты для переодевания следует проектировать и использовать как воздушные шлюзы и оборудовать, при необходимости, умывальниками и душевыми кабинами. Схема перепада давления воздуха должна предотвращать движение воздуха между рабочей зоной и окружающей средой, а также риск контаминации одежды, используемой вне рабочей зоны;
f) для перемещения оборудования должна быть предусмотрена система воздушных шлюзов, конструкция которой должна предотвращать движение контаминированного воздуха рабочей зоны во внешнюю среду и риск контаминации оборудования внутри шлюза. Размер шлюза должен позволять проводить эффективную деконтаминацию поверхности перемещаемых материалов. Рекомендуется устанавливать на двери шлюза таймер, позволяющий контролировать время, достаточное для проведения эффективной деконтаминации;
g) во многих случаях для безопасного удаления отходов и подачи стерильных предметов необходимо использовать проходной автоклав с двумя дверями.
12. Передаточные шлюзы и комнаты для переодевания следует оборудовать блокирующими или другими подходящими устройствами, препятствующими одновременному открыванию более чем одной двери. В комнаты для переодевания следует подавать отфильтрованный воздух той же степени очистки, что и для производственных зон. Комнаты для передевания должны быть оборудованы системами удаления воздуха, обеспечивающими воздухообмен, независимый от производственных зон. Передаточные шлюзы должны, как правило, вентилироваться таким же образом, однако допускается применять невентилируемые шлюзы или шлюзы, оборудованные только приточными системами.
13. Стадии технологического процесса, при которых может произойти контаминация продукции (хранение клеток, приготовление сред, культивирование вирусов и др.), должны выполняться в отдельных зонах. Особая предосторожность требуется при работе с животными и продуктами животного происхождения.
14. Операции с микроорганизмами, проявляющими высокую устойчивость к дезинфекции (например, спорообразующие бактерии), до их инактивации следует выполнять в изолированных зонах, специально предназначенных для проведения таких операций.
15. В производственной зоне следует одновременно проводить работы с микроорганизмами только одного вида, за исключением процессов смешивания и последующего наполнения.
16. Следует проектировать производственные зоны таким образом, чтобы предусмотреть возможность проведения дезинфекции производственных зон в промежутках между производственными циклами с использованием валидированных методов.
17. Допускается производство микроорганизмов в контролируемых зонах при условии, что оно осуществляется в полностью закрытом и термически стерилизуемом оборудовании, а все соединения после завершения работы и перед разборкой также подвергаются термической стерилизации. Допускается выполнение соединений под локальным ламинарным потоком воздуха при условии, что их количество ограничено, используются соответствующие асептические методы и отсутствует опасность утечки. Параметры процесса стерилизации, используемого перед разборкой соединений, должны пройти валидацию для всех используемых микроорганизмов. В пределах одной зоны разные продукты могут быть загружены в разные биореакторы только при отсутствии риска случайной перекрестной контаминации. Однако работа с микроорганизмами, к которым предъявляются особые требования изоляции, должна выполняться только в зонах, специально выделенных для такой продукции.
18. В помещениях для содержания животных, предназначенных для использования (или используемых) в производстве, следует поддерживать режим изолированной и (или) чистой зоны, также помещения следует отделять от помещений для содержания других животных. Помещения, где содержатся животные, используемые для контроля качества продукции (в том числе с применением патогенных микроорганизмов), должны быть соответствующим образом изолированы.
19. Доступ в производственные зоны может иметь только персонал, имеющий на это разрешение. Необходимо иметь однозначную и лаконичную письменную инструкцию по режиму доступа в производственные зоны, которую рекомендуется разместить на стене.
20. Техническая документация (основной файл) предприятия должна содержать необходимые данные о производственных помещениях.
Должна быть обеспечена соответствующая детализация при описании производственных участков и зданий (путем включения планов помещений и их экспликации), назначения и условий использования всех помещений, а также видов биологических агентов, с которыми проводится работа. Должны быть четко обозначены также потоки движения персонала и продукции. Необходимо указать виды животных, содержащихся в вивариях или других помещениях предприятия, а также виды работ, выполняемых вблизи производственного участка.
Планы изолированных и (или) чистых помещений и зон должны содержать принципиальную схему вентиляции с указанием мест притока и вытяжки воздуха, используемых фильтров и их спецификаций, кратностей воздухообмена и перепадов давления. Следует указать, между какими помещениями предусмотрен мониторинг перепада давления с помощью манометров.
Оборудование
21. Используемое оборудование должно быть сконструировано и смонтировано так, чтобы соответствовать специфическим требованиям для каждого вида продукции.
Перед вводом оборудования в эксплуатацию оно должно пройти квалификацию и валидацию, а затем проходить регулярное техническое обслуживание и повторную валидацию.
22. Оборудование должно обеспечивать удовлетворительную первичную изоляцию биологических агентов. Конструкция и монтаж оборудования должны, при необходимости, обеспечивать его легкую и эффективную деконтаминацию и (или) стерилизацию.
23. Конструкция и монтаж закрытого оборудования, используемого для первичной изоляции биологических агентов, должны предусматривать предотвращение утечек, образования капель и аэрозолей.
Вводы и выводы газов должны быть защищены так, чтобы обеспечивать нужную степень изоляции, например, путем использования стерилизующих гидрофобных фильтров.
Для подачи или удаления материалов следует использовать стерилизуемую закрытую систему либо соответствующие условия ламинарного потока воздуха.
24. При необходимости перед использованием оборудование следует стерилизовать предпочтительно сухим паром под давлением. Другие методы применимы в случае, если свойства оборудования не допускают его стерилизацию паром. Важно не пропустить при обработке такие виды оборудования, как центрифуги и водяные бани.
Оборудование, используемое для очистки, разделения или концентрирования, должно как минимум стерилизоваться или дезинфицироваться при переходе от одного вида продукта к другому. Необходимо исследовать влияние методов стерилизации на эффективность и валидационный статус оборудования с целью определения срока его эксплуатации.
Все методы стерилизации должны быть валидированы.
25. Конструкция оборудования должна исключать возможность перепутывания различных организмов или продуктов. Трубопроводы, клапаны и фильтры должны маркироваться в соответствии с их назначением.
Для контейнеров с инфицированными и неинфицированными материалами, а также, как правило, для различных организмов или клеток следует использовать отдельные инкубаторы. Содержание в одном инкубаторе более 1 типа организмов или типа клеток допускается только в случае принятия необходимых мер по герметизации, поверхностной деконтаминации и разделению контейнеров. Сосуды с культурами и др. должны быть промаркированы индивидуально. Очистка и дезинфекция этих сосудов и инкубаторов могут быть затруднены и требовать особого внимания.
Конструкция и порядок эксплуатации оборудования, используемого для хранения биологических агентов или продуктов, должны исключать любое возможное перепутывание.
Все хранящиеся образцы должны иметь четкую и однозначную маркировку и находиться в контейнерах, защищенных от утечки. Запасы посевных культур клеток и организмов должны храниться в специально предназначенном оборудовании.
26. Некоторые виды оборудования (например, оборудование, требующее контроля температуры) должны быть оснащены регистрирующими устройствами и (или) системами сигнализации. Во избежание отказов совместно с анализом тенденций в регистрируемых данных следует организовать систему профилактического обслуживания.
27. Загрузка лиофильных сушильных установок должна происходить в чистой (изолированной) зоне. Операции по разгрузке лиофильной сушильной установки приводят к загрязнению окружающей среды. В связи с этим при использовании лиофильных сушильных установок, открывающихся с одной стороны, чистое помещение должно пройти деконтаминацию до перемещения в эту зону другой серии продукции, за исключением серий с одинаковыми организмами. Двусторонние лиофильные сушильные установки должны стерилизоваться после каждого цикла производства, если только они не открываются в чистую зону.
Стерилизация лиофильных сушильных установок должна проводиться в соответствии с пунктом 24 настоящих Требований. В случае организации работ по принципу отдельных циклов производства их следует стерилизовать, по крайней мере, после каждого цикла.
Животные и виварии
28. Общие требования к содержанию животных, помещениям для них, уходу и карантину устанавливаются нормативными правовыми актами государств - членов Евразийского экономического Союза.
29. Виварии должны быть изолированы от других производственных помещений. Они должны быть сконструированы с соблюдением необходимых требований.
30. Санитарное состояние животных, используемых в производстве, необходимо оценивать, контролировать и регистрировать. Правила обращения с некоторыми видами животных (например, линии животных, свободных от патогенов) устанавливаются в специальных руководствах или нормативных правовых актах государств - членов Евразийского экономического союза.
31. Животные, биологические агенты и проводимые испытания должны быть четко идентифицированы в соответствии с установленной системой во избежание риска перепутывания и с целью контроля всех возможных видов опасностей.
Дезинфекция.
Удаление отходов
32. Дезинфекция и (или) удаление твердых и жидких отходов могут иметь особенно важное значение при производстве иммунобиологической продукции. В связи с этим необходим тщательный подход к методам и оборудованию, используемым для предотвращения загрязнения окружающей среды, а также к их валидации.
Производство
33. Из-за широкого разнообразия продукции, большого количества стадий при производстве иммунобиологических ветеринарных лекарственных средств и характера биологических процессов особое внимание следует уделять строгому соблюдению валидированных технологических процессов, постоянному контролю всех технологических стадий производства и проведению контроля в процессе производства.
Особое внимание следует обратить на исходное сырье, материалы, среды культивирования и использование систем посевных культур.
Исходное сырье и материалы
34. Требования к исходному сырью и материалам должны быть четко определены в документально оформленных спецификациях. Спецификации должны содержать данные о поставщике, методе производства, месте расположения поставщика и видах животных, из которых получено исходное сырье и материалы, а также о способах его контроля. Особенную роль играют микробиологические методы контроля.
35. Результаты испытаний исходного сырья и материалов должны соответствовать спецификациям. Если проведение испытаний занимает длительный период времени (например, свободные от специфических патогенов (SPF) яйца (эмбрионы) птиц), может возникнуть необходимость обработки исходного сырья и материалов до получения результатов аналитических испытаний. В этих случаях получение разрешения на выпуск готовой продукции зависит от положительных результатов испытания исходного сырья и материалов.
36. Особое внимание следует уделять данным о системе обеспечения качества поставщика при оценке соответствия источников сырья и материалов и объеме испытаний, требуемых при проведении входного контроля качества.
37. По возможности следует стерилизовать исходное сырье и материалы термическим методом. Допускается использовать и другие валидированные методы, например, ионизирующее излучение.
Среды
38. Ростовые свойства сред должны заранее подтверждаться соответствующим образом.
39. Предпочтительно, чтобы среды стерилизовались на месте или на производственной линии. Предпочтительным является метод термической стерилизации. Газы, среды, кислоты, щелочи, пеногасители и другие вещества, вводимые в стерильный биореактор, должны быть стерильными.
Система посевных культур и банков клеток
40. Для предотвращения нежелательного изменения свойств, которое может произойти вследствие многократных пересевов или большого числа генераций, производство иммунобиологических ветеринарных лекарственных средств, получаемых из культур микроорганизмов, культур клеток, тканей или размножением в эмбрионах и животных, должно быть основано на системе посевных культур или банков клеток.
41. Количество генераций (удвоений, пассажей) между посевной культурой или банком клеток и готовой продукцией должно соответствовать требованиям регистрационного досье.
42. Посевные культуры и банки клеток следует соответствующим образом различать и контролировать на отсутствие контаминантов. Для новых посевных культур должны быть установлены критерии приемлемости. Посевные культуры и банки клеток должны создаваться, содержаться и использоваться таким образом, чтобы свести к минимуму риск их контаминации или любого изменения. При создании посевных культур или банков клеток не допускается одновременная работа в той же зоне или того же персонала с другими живыми или инфицирующими материалами (например, вирусами или клеточными линиями).
43. Создание посевной культуры или банка клеток необходимо проводить с соблюдением установленных требований к производственной среде для защиты посевных культур и банка клеток, а также работающего с ними персонала и внешней окружающей среды.
44. Происхождение, форма и условия хранения посевных культур должны быть полностью описаны в соответствующей документации. Необходимо иметь доказательства стабильности и воспроизводимости посевных культур и клеток. Емкости для хранения должны быть герметично закрыты, четко маркированы и храниться при требуемой температуре. Условия хранения необходимо должным образом контролировать. Необходимо вести тщательный учет каждого хранящегося контейнера.
45. К работе с материалом допускаются только специально назначенные сотрудники. Работа должна проводиться под контролем ответственного лица. Различные посевные культуры и банки клеток следует хранить таким образом, чтобы не допускать перепутывания или перекрестной контаминации. Рекомендуется разделять посевные культуры и банки клеток и их разные части хранить отдельно во избежание их полной утраты.
Принципы работы
46. В ходе технологических процессов следует избегать или сводить к минимуму образование капель или пены. Процессы центрифугирования или смешивания, которые могут привести к образованию капель, должны проводиться в изолированных емкостях или в чистых (изолированных) зонах во избежание переноса живых организмов.
47. Случайные проливы жидкостей, особенно содержащих живые организмы, необходимо быстро и безопасным способом ликвидировать. Для каждого типа микроорганизмов следует использовать валидированные процедуры деконтаминации. При использовании различных штаммов бактерий одного вида или очень похожих вирусов эта процедура может быть валидирована в отношении только одного из них, если у них нет существенных с испытанным агентом различий в устойчивости к воздействию деконтаминации.
48. Операции, включающие в себя перемещение таких материалов, как стерильные среды, культуры или продукты, должны проводиться в предварительно стерилизованных закрытых системах. Если это невозможно, операции по перемещению материалов должны проводиться в ламинарном потоке воздуха.
49. Добавление сред или культур в биореакторы и другие сосуды должно проводиться в тщательно контролируемых условиях, обеспечивающих невозможность внесения контаминантов. При добавлении культур необходимо тщательно проверять правильность соединения сосудов.
50. При необходимости (например, когда 2 и более ферментатора расположены в 1 зоне) отверстия для отбора проб и внесения добавок и соединительные элементы должны стерилизоваться паром (после подсоединения, перед подачей продукта и перед отсоединением). В других случаях допускаются химическая дезинфекция входных отверстий и защита соединений под ламинарным потоком воздуха.
51. Оборудование, лабораторная посуда, внешние поверхности контейнеров с продукцией и другие подобные материалы перед перемещением из изолированной зоны должны быть дезинфицированы с использованием валидированного метода (в соответствии с требованиями, указанными в пункте 47 настоящего документа). Особую проблему может вызвать ведение документации на серию продукции. Только абсолютный минимум документации, необходимой для соблюдения требований Правил, должен поступать в рабочую зону и покидать ее. При явной контаминации (например, каплями или аэрозолями) или при использовании экзотических микроорганизмов бумажная документация должна дезинфицироваться в шлюзе для перемещения оборудования или передаваться с использованием фотокопии или факса.
52. Жидкие или твердые отходы, такие как скорлупа после отбора материала из яиц, одноразовые бутыли для культур, нежелательные культуры или биологические агенты целесообразно стерилизовать или дезинфицировать перед их удалением из изолированной зоны. В некоторых случаях приемлемыми считаются и другие методы (например, использование герметичных контейнеров или трубопроводов).
53. Необходимо осуществлять тщательный контроль за тем, чтобы в производственную зону попадали только те предметы, исходное сырье и материалы, в том числе документация, которые относятся к производимой продукции. Для контроля за соответствием количества вносимых и выносимых предметов и материалов во избежание их накопления в производственном помещении должна действовать система учета.
54. Термостойкие предметы и материалы должны поступать в чистую или чистую изолированную зону через проходные автоклавы или сухожаровые печи. Чувствительные к нагреву предметы и материалы должны поступать через воздушный шлюз с блокируемыми дверями, где эти предметы и материалы подвергаются дезинфекции. Допускается стерилизация предметов и материалов в другом месте, если они поступают через шлюз в двойной оболочке с соблюдением необходимых мер предосторожности.
55. Во избежание контаминации или перепутывания культур клеток или микроорганизмов на протяжении периода инкубации необходимо принимать меры предосторожности. Должна существовать методика очистки и дезинфекции инкубаторов. Контейнеры, находящиеся в инкубаторах, должны быть четко и тщательно маркированы.
56. В одном помещении допускается одновременное проведение операций только с 1 живым биологическим агентом, за исключением операций по смешиванию и последующей фасовке или при использовании полностью закрытых систем. В промежутках между операциями с различными живыми биологическими агентами производственные помещения должны проходить эффективную дезинфекцию.
57. Продукты должны инактивироваться путем добавления инактиватора с последующим тщательным перемешиванием. После этого смесь должна переноситься во 2-й стерильный сосуд, за исключением случаев, когда форма и размер сосуда позволяют его легко переворачивать и встряхивать таким образом, чтобы все внутренние поверхности смачивались конечной смесью культур и инактиватора.
58. Сосуды, содержащие инактивированный продукт, не допускается открывать, из этих сосудов также не допускается отбирать пробы в зонах, где содержатся живые биологические агенты. Всю последующую обработку инактивированных продуктов следует проводить в чистых зонах классов A и B или в закрытом оборудовании, специально предназначенном для инактивированных продуктов.
59. Необходимо уделять особое внимание валидации методов стерилизации, дезинфекции, удаления и инактивации вирусов.
60. Наполнение следует проводить в как можно более короткий промежуток времени после завершения производственных операций. До начала операции наполнения емкости с нерасфасованной продукцией должны быть герметично закрыты, соответствующим образом маркированы и храниться при установленных температурных условиях.
61. Должна существовать система, обеспечивающая контроль целостности и герметичности упаковок после их наполнения.
62. Флаконы, содержащие живые биологические агенты, должны быть закрыты крышками таким образом, чтобы обеспечивалась невозможность контаминации другой продукции или проникновение живых агентов в другие зоны или во внешнюю среду.
63. Между наполнением первичных упаковок, их маркировкой и упаковкой может пройти определенный промежуток времени. Должны быть разработаны и документально оформлены процедуры, описывающие порядок хранения немаркированных контейнеров, обеспечивающие невозможность их перепутывания и надлежащие условия хранения. Особое внимание следует уделять хранению термолабильной и светочувствительной продукции. Температура хранения должна быть установлена.
64. На каждой технологической стадии следует проводить сопоставление фактического и ожидаемого выхода продукции. Все существенные отклонения должны быть расследованы.
Контроль качества
65. В обеспечении стабильности качества иммунобиологических лекарственных средств особое значение имеет контроль в процессе производства. Виды контроля, которые имеют решающее значение для оценки качества (например, контроль на отсутствие вирусов), но не могут быть проведены на готовой продукции, должны выполняться на одной из предшествующих стадий производства.
66. Для повторения или подтверждения результатов контроля качества серии продукции может возникнуть необходимость хранения достаточного объема образцов промежуточных продуктов при соответствующих условиях.
67. Может возникнуть необходимость непрерывного контроля параметров в ходе процесса производства (например, непрерывного контроля физических параметров в ходе ферментации).
68. Распространенной практикой является непрерывное культивирование биологической продукции. При таком методе производства необходимо принять во внимание особые требования к организации контроля качества продукции.
ПРИЛОЖЕНИЕ № 6
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству медицинских газов
Принцип
Производство медицинских газов, относящихся к лекарственным средствам, следует осуществлять в соответствии с законодательством государств - членов Евразийского экономического союза и актами органов Евразийского экономического союза. Настоящий документ устанавливает требования к производству газов как активных фармацевтических субстанций (далее - АФС) и производству газов для медицинского применения.
Разграничение между производством АФС и лекарственных препаратов (медицинских газов) должно быть четко определено в каждом регистрационном досье. Обычно стадии производства и очистки газов относят к производству АФС. К производству лекарственных препаратов относят газы, поступающие на последующие этапы производства из контейнеров первичного хранения.
Производство газов как АФС следует осуществлять в соответствии с требованиями, указанными в разделе II Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила), настоящим документом и другими соответствующими приложениями к Правилам.
Производство лекарственных препаратов (медицинских газов) следует осуществлять в соответствии с требованиями, указанными в части I Правил, настоящим документом и другими соответствующими приложениями к Правилам.
В исключительных случаях непрерывные процессы, начиная с исходного сырья для производства АФС до производства лекарственного препарата, при которых невозможно промежуточное хранение газа, следует рассматривать как производство лекарственных препаратов. Это должно быть четко указано в регистрационном досье.
Положения настоящего документа не распространяются на производство и обращение медицинских газов в медицинских учреждениях, если такой процесс не является промышленным производством. Тем не менее, соответствующие разделы настоящего документа могут быть использованы в качестве основы для организации этой работы.
Производство газов как активных фармацевтических субстанций
Газы как активные фармацевтические субстанции могут быть получены путем химического синтеза или из натуральных источников путем их очистки (при необходимости) (например, на заводах по разделению воздуха).
1. Технологические процессы получения газов этими двумя методами следует осуществлять в соответствии с требованиями, указанными в части II Правил, с учетом следующего:
а) требования к исходному сырью для активных фармацевтических субстанций (раздел 7 части II Правил) не применимы к производству активных фармацевтических субстанций (далее - АФС) - газов методом разделения воздуха (однако производитель должен гарантировать, что качество используемого воздуха соответствует установленному и изменения качества воздуха из внешней среды не будут оказывать влияние на качество газов, производимых как АФС);
b) требования к непрерывному изучению стабильности активных фармацевтических субстанций (пункт 11.5 части II Правил), осуществляемому с целью подтверждения условий хранения и срока годности (или даты повторного контроля) (пункт 11.2 части II Правил) не применяются в случае, если в качестве данных первичного изучения стабильности были использованы литературные данные;
c) требования к контрольным и архивным образцам ( пункт 11.7 части II Правил) не применяются к газам как активным фармацевтическим субстанциям, если иное не установлено.
2. Следует проводить постоянный мониторинг качества газов как активных фармацевтических субстанций, производимых непрерывным методом (например, разделением воздуха). Результаты мониторинга следует хранить в виде, позволяющем осуществлять оценку тенденций.
3. Кроме того:
a) условия транспортирования и доставки нерасфасованных газов как активных фармацевтических субстанций должны соответствовать требованиям, установленным для медицинских газов ( пункты 19 - 21 настоящего документа);
b) операции наполнения баллонов или переносных криогенных емкостей газами как активными фармацевтическими субстанциями следует проводить в соответствии с требованиями, установленными для медицинских газов (пункты 22 - 37 настоящего документа), и требованиями раздела 9 части II Правил.
Производство медицинских газов
Производство медицинских газов, как правило, осуществляется в закрытом оборудовании. В связи с этим риск контаминации данной продукции из производственной среды является минимальным. Однако существует риск контаминации (или перекрестной контаминации другими газами) в особенности при повторном использовании емкостей.
4. Требования, применяемые к баллонам, следует также применять к группам (связкам) баллонов (за исключением случев хранения и транспортирования в специальных контейнерах).
Персонал
5. Персонал, занятый в производстве и реализации медицинских газов, должен пройти соответствующее обучение требованиям Правилам, специфическим в отношении данного вида продукции. Персонал должен знать критически важные аспекты и возможную опасность, которую лекарственные препараты в виде медицинских газов могут представлять для пациентов. Водители, осуществляющие перевозку медицинских газов, также должны пройти соответствующее обучение.
6. Персонал подрядчиков, который может оказать влияние на качество медицинских газов (например, персонал, осуществляющий техническое обслуживание баллонов или клапанов), должен пройти соответствующее обучение.
Помещения и оборудование
Помещения
7. Баллоны и переносные криогенные емкости необходимо проверять, подготавливать, наполнять и хранить в зонах, отделенных от зон работы с немедицинскими газами. Запрещается обмен баллонами (переносными криогенными емкостями) между этими зонами. Однако в одних и тех же зонах допускаются контроль, подготовка, наполнение и хранение других газов, если требования к ним соответствуют требованиям к медицинским газам, а производственные операции осуществляются в соответствии с требованиями Правил.
8. Помещения, в которых выполняются операции по производству, проведению испытаний и хранению медицинских газов, должны иметь площадь, достаточную для исключения риска перепутывания. Планировка помещений должна обеспечивать:
a) отдельные маркированные зоны для различных газов;
b) однозначное обозначение и разделение пустых баллонов (переносных криогенных емкостей) и баллонов (переносных криогенных емкостей), находящихся на разных стадиях производства (например, надписи: «ожидает контроля», «ожидает наполнения», «карантин», «разрешен», «отклонен», «готов к отправке»).
Способ, используемый для достижения различных уровней разделения, зависит от характера, объема и сложности технологического процесса. Мерами по разделению могут быть разметка по зонам на полу, перегородки, барьеры, обозначения и др.
9. После сортировки или технического обслуживания пустые баллоны (переносные криогенные емкости) и наполненные баллоны (переносные криогенные емкости) следует хранить под навесами, защищающими их от неблагоприятных погодных условий. Наполненные баллоны (переносные криогенные емкости) следует хранить в условиях, гарантирующих их доставку в чистом виде, который отвечает условиям, в которых они будут использоваться.
10. Должны обеспечиваться особые условия хранения, соответствующие требованиям регистрационного досье (например, для газовых смесей, в которых происходит разделение фаз в случае замораживания).
Оборудование
11. Оборудование должно быть спроектировано таким образом, чтобы гарантировать, что надлежащий газ наполняется в надлежащий контейнер. Как правило, не должно быть соединений между трубопроводами, по которым проходят различные газы. В случае, если такие соединения необходимы (например, оборудование, используемое для наполнения смесями газов), путем проведения квалификации необходимо гарантировать отсутствие риска перекрестной контаминации разными газами. В дополнение к этому распределительные коллекторы должны быть оборудованы специфическими соединительными элементами. Требования к использованию распределительных коллекторов и их соединений с клапанами баллонов могут быть установлены в соответствующих нормативных правовых актах. Использование на одном производственном участке соединений, соответствующих различным стандартам, следует тщательно контролировать, также, как и использование адаптеров, применение которых бывает необходимым в некоторых случаях соединения специфических систем наполнения.
12. Резервуары для хранения и передвижные цистерны для доставки должны быть предназначены только для одного вида газа определенного качества. Медицинские газы могут храниться или транспортироваться в тех же резервуарах, что и аналогичные немедицинские газы, при условии, что качество последних по меньшей мере эквивалентно качеству медицинских газов и соблюдаются требования Правил. В таких случаях должна быть осуществлена и документально оформлена процедура управления рисками.
13. Общая система распределения газа с коллекторами медицинского и немедицинского назначения используется лишь при наличии валидированного метода недопущения обратного потока газа из немедицинской системы в медицинскую систему.
14. Должны быть специально предназначенные коллекторы наполнения для одного медицинского газа или определенной смеси медицинских газов. В исключительных случаях допускается наполнение газов для других медицинских целей с использованием коллекторов, предназначенных для медицинских газов, если доказана такая возможность и весь процесс находится под контролем. В этих случаях качество немедицинского газа должно быть по крайней мере равным требуемому качеству медицинского газа и должны соблюдаться требования Правил. Наполнение должно производиться по принципу организации циклов производства.
15. Работы по ремонту и техническому обслуживанию оборудования (включая очистку и продувку оборудования) не должны влиять на качество медицинских газов. В частности следует разработать и документально оформить мероприятия, проводимые после ремонта и технического обслуживания оборудования, включающих разгерметизацию систем. Особенно важным является подтверждение того, что оборудование свободно от любой контаминации, которая может оказать влияние на качество готового продукта до выпуска его в реализацию. Записи проведенных работ должны сохраняться.
16. Следует разработать и документально оформить процедуры, осуществляемые при возврате цистерны на участок работы с медицинскими газами (после транспортирования немедицинского газа в условиях, указанных в пункте 12 настоящего документа, или после операций по техническому обслуживанию). Такая процедура должна включать в себя аналитические испытания на наличие остатков продукта.
Документация
17. Данные, включенные в досье на каждую серию наполненных баллонов (переносных криогенных емкостей), должны обеспечивать для каждого наполненного баллона прослеживаемость информации о всех основных параметрах соответствующих стадий наполнения. Записи, относящиеся к серии (досье на серию), должны, как правило, содержать следующую информацию:
a) наименование продукции;
b) номер серии;
c) дата и время проведения операции по наполнению;
d) персонал, выполнявший существенные этапы процесса (например, очистку линий, получение исходного сырья и материалов, подготовку линий наполнения, проведение наполнения и др.);
e) ссылка на номер серии газа, который использовался для операций по наполнению в соответствии с пунктом 22настоящего документа, включая его статус (разрешение к наполнению);
f) оборудование, которое использовалось (например, коллектор наполнения);
g) количество баллонов (переносных криогенных емкостей) до операции наполнения, включая идентификационные данные каждой емкости и ее геометрический объем;
h) операции, выполненные до наполнения (в соответствии с пунктом 30 настоящего документа);
i) ключевые параметры, необходимые для подтверждения правильности проведения операции по наполнению при стандартных условиях;
j) результаты соответствующих проверок, гарантирующих, что баллоны (переносные криогенные емкости) были наполнены;
k) образец этикетки серии;
l) спецификация лекарственного средства и результаты испытаний по контролю качества (включая ссылки на текущее состояние калибровки оборудования, использованного в ходе испытаний);
m) количество отклоненных баллонов (переносных криогенных емкостей) с указанием их индивидуальных идентификационных данных и причины отклонения;
n) подробные сведения о всех проблемах и отклонениях, утвержденное разрешение на любое отклонение от инструкций по наполнению;
о) разрешение уполномоченного лица на выпуск серии, дата и его подпись.
18. Должны сохраняться досье на каждую серию газа, предназначенную для наполнения резервуаров в медицинских учреждениях. Требования к содержанию таких досье зависят от требований соответствующих нормативных правовых актов государств - членов Евразийского экономического союза, но, как правило, досье должно включать в себя следующую информацию:
a) наименование продукции;
b) номер серии;
c) ссылка на идентификационный номер емкости (цистерны), в которой серия разрешена к реализации;
d) дата и время операции по наполнению;
e) персонал, выполнивший наполнение емкости (цистерны);
f) информация о контейнере, из которого производилось наполнение, и о газе, использовавшемся для наполнения;
g) сведения обо всех существенных деталях наполнения;
h) спецификация на готовый лекарственный препарат и результаты контроля качества (включая ссылки на текущее состояние калибровки оборудования, использованного в ходе испытаний);
i) подробные сведения обо всех проблемах и отклонениях, утвержденное разрешение на любое отклонение от инструкций по наполнению;
j) разрешение уполномоченного лица на выпуск серии, дата и его подпись.
Производство
Перемещение и доставка криогенных и сжиженных газов
19. Перемещение криогенных или сжиженных газов с места первичного хранения (включая контроль перед перемещением) следует осуществлять в соответствии с валидированными процедурами, разработанными с целью предотвращения возможной контаминации. Трубопровод, по которому перемещается газ, должен быть оборудован обратным клапаном или другим соответствующим устройством. Гибкие соединения, нестационарные соединительные шланги и средства для соединения перед использованием должны быть промыты потоком соответствующего газа.
20. Шланги, используемые для наполнения резервуаров и цистерн, должны быть оборудованы средствами для соединения, специально предназначенными для данной продукции. Использование адаптеров, позволяющих подключать резервуары и цистерны, должно контролироваться надлежащим образом.
21. Подача газа в резервуары, содержащие аналогичный газ такого же уровня качества, может быть осуществлена при наличии положительных результатов испытаний качества подаваемого газа. Испытуемый образец может быть отобран как из подаваемого газа, так и из резервуара после завершения подачи газа.
Особые меры в отношении наполнения резервуаров, расположенных у потребителей, изложены в пункте 42 настоящего документа.
Наполнение и маркировка баллонов (переносных криогенных емкостей)
22. Перед наполнением баллонов (переносных криогенных емкостей) серия (серии) газа должна быть идентифицирована и проконтролирована в соответствии со спецификациями и разрешена для проведения наполнения.
23. В случае непрерывных процессов, определенных в принципах настоящего документа, для обеспечения соответствия газа спецификациям следует установить соответствующие точки контроля производства.
24. Баллоны, переносные криогенные емкости и клапаны должны отвечать установленным техническим спецификациям и требованиям регистрационного досье. Они должны быть предназначены только для одного медицинского газа или определенной смеси медицинских газов. Баллоны должны быть маркированы с использованием цветовой маркировки по соответствующим стандартам. Для обеспечения соответствующей защиты от контаминации баллоны следует оснащать клапанами удержания минимального давления с механизмами предотвращения потока газа в обратном направлении.
25. Баллоны, переносные криогенные емкости и клапаны следует проверять перед первым использованием в производстве и следует обслуживать надлежащим образом. При использовании медицинских изделий их техническое обслуживание должно осуществляться в соответствии с инструкциями производителя.
26. Операции по проверке и техническому обслуживанию не должны оказывать негативного воздействия на качество и безопасность лекарственного препарата. Вода, используемая для испытаний баллонов гидростатическим давлением, должна быть как минимум питьевого качества.
27. Для подтверждения отсутствия контаминации внутреннее состояние баллонов до установки клапана должно подвергаться визуальному осмотру на предмет отсутствия остатков воды или других контаминантов. Эту операцию следует выполнять как часть проверок и технического обслуживания. Она должна осуществляться в следующих случаях:
новые баллоны впервые используются для медицинских газов;
были проведены испытания гидростатическим давлением или эквивалентным испытанием с демонтажем клапана;
произведена замена клапана.
После установки клапан должен находиться в закрытом состоянии для предотвращения любой контаминации. В случае возникновения любых сомнений относительно внутреннего состояния баллона клапан должен быть демонтирован, а баллон подвергнут внутреннему осмотру для обеспечения уверенности в отсутствии контаминации.
28. Производитель лекарственных препаратов несет ответственность за техническое обслуживание и ремонт баллонов, переносных криогенных емкостей и клапанов. При выполнении этих работ по соглашению они должны выполняться только утвержденными исполнителями. Соглашение должно содержать технические условия выполнения таких работ. Следует проводить аудит исполнителей таких работ для обеспечения уверенности в соблюдении ими соответствующих стандартов.
29. Должна быть в наличии система, позволяющая обеспечить прослеживаемость баллонов, переносных криогенных емкостей и клапанов.
30. Проверка перед операцией наполнения должна включать в себя:
а) в случае баллонов - проверку по установленной процедуре наличия остаточного избыточного давления для каждого баллона:
если баллон оборудован клапаном удержания минимального давления, при отсутствии сигнала, свидетельствующего о наличии остаточного избыточного давления, должна быть проведена проверка клапана (если клапан функционирует неправильно, баллон должен быть отправлен на техническое обслуживание);
если баллон не оборудован клапаном удержания минимального давления и в баллоне не обнаружено остаточного избыточного давления, такой баллон должен быть отправлен для проведения дополнительных испытаний с целью проверки отсутствия контаминации водой или другими веществами (дополнительные меры могут включать в себя визуальный осмотр внутреннего состояния баллона, который проводится после очистки с использованием валидированного метода);
b) проверку для подтверждения того, что идентификационные этикетки предыдущей серии отсутствуют;
c) проверку того, что все поврежденные идентификационные этикетки продукта удалены и заменены;
d) внешний визуальный осмотр каждого баллона, переносной криогенной емкости и клапана с целью выявления раковин, сварочных прожигов, других повреждений, контаминации маслами и при необходимости проведение очистки;
e) проверку соединения патрубка каждого баллона или переносной криогенной емкости на соответствие типу соединения для наполняемого газа;
f) проверку даты следующего испытания клапана (для клапанов, подлежащих периодической проверке);
g) проверку баллонов или переносных криогенных емкостей для обеспечения гарантии проведения всех необходимых испытаний (например, проверку гидростатическим давлением или эквивалентным испытанием) в соответствии с нормативными правовыми актами государств - членов Евразийского экономического союза и проверку действительности результатов этих испытаний;
h) проверку цветовой маркировки каждого баллона в соответствии с регистрационным досье (цветовая кодировка устанавливается нормативными правовыми актами государств - членов Евразийского экономического союза).
31. Размер серии должен быть определен в зависимости от операции наполнения.
32. Баллоны, возвращаемые на повторную заправку, должны быть тщательно подготовлены с целью минимизации риска контаминации согласно требованиям регистрационного досье. Методики, включающие в себя процедуры откачивания и (или) продувки, должны быть валидированы.
Для сжатых газов теоретическое содержание примеси при давлении наполнения 200 бар должно составлять не более 500 объемных частей на 1 000 000 при 15 0 C. Для других давлений определяются эквивалентные значения.
33. С целью минимизации риска контаминации переносные криогенные емкости, возвращаемые на повторную заправку, должны тщательно подготавливаться согласно процедурам, описанным в регистрационном досье. В частности переносные емкости, в которых отсутствует остаточное давление, должны быть подготовлены с использованием валидированного метода.
34. Для обеспечения гарантии правильного наполнения каждого баллона (переносной криогенной емкости) следует проводить соответствующие проверки.
35. Каждый наполненный баллон до установки устройства контроля первого вскрытия (пункт 36 настоящего документа) должен быть проконтролирован на отсутствие утечки с использованием соответствующего метода. Используемый метод контроля не должен приводить к контаминации поверхности патрубка клапана баллона, и по возможности такой контроль должен осуществляться после отбора всех образцов для контроля качества.
36. После наполнения газом патрубки клапанов баллонов должны быть закрыты колпачками для защиты от контаминации. На баллоны (переносные криогенные емкости) должны быть установлены устройства контроля первого вскрытия.
37. Каждый баллон (переносная криогенная емкость) должен быть промаркирован с помощью этикеток. Номер серии и срок годности могут быть указаны на отдельной этикетке.
38. При производстве медицинских газов путем смешивания двух или более различных газов (в линии для наполнения либо непосредственно в баллонах) должен использоваться валидированный метод смешивания, который гарантирует, что газы должным образом смешаны в каждом баллоне и гомогенность смеси обеспечена.
Контроль качества
39. Каждая серия медицинского газа (баллоны, переносные криогенные емкости, резервуары в медицинских учреждениях) должна быть проконтролирована в соответствии с регистрационным досье и на каждую серию медицинского газа должно быть получено разрешение уполномоченного лица на выпуск.
40. План отбора проб и объем проводимых испытаний должны отвечать следующим требованиям в отношении баллонов (если в регистрационном досье не установлено иное):
а) если только один медицинский газ наполняется в баллоны с использованием коллектора, к которому одновременно подсоединяются несколько баллонов, газ должен быть проконтролирован как минимум из одного баллона для установления подлинности и количественного определения. Образцы должны отбираться от каждого цикла наполнения при замене баллонов, подключенных к коллектору;
b) если только один медицинский газ наполняется в баллоны в рамках одного производственного цикла в один промежуток времени, по крайней мере один баллон при каждом непрерывном цикле наполнения должен быть проверен на подлинность и количественное содержание. Примером непрерывного цикла наполнения является производство в течение одной смены одним и тем же персоналом с использованием одного оборудования и одной серии газа, который расфасовывался;
c) если медицинский газ готовится путем смешивания в баллоне двух или более различных газов из одного и того же распределительного коллектора, газ из каждого баллона должен быть проконтролирован на подлинность и количественное содержание всех компонентов газовой смеси. В отношении вспомогательных веществ (при их наличии) испытание на подлинность может выполняться для одного баллона из цикла наполнения (или для каждого непрерывного цикла наполнения). Меньшее количество баллонов может подвергаться испытаниям в случае использования валидированных автоматизированных систем наполнения;
d) в отношении газов, смешивание которых происходит до наполнения, следует соблюдать те же принципы, что и в отношении наполнения одним газом, когда на линии осуществляется непрерывный контроль смеси газов, используемых для наполнения.
В отношении газов, смешивание которых происходит до стадии наполнения, следует придерживаться тех же принципов, как и для газов, смешивание которых проводится в баллонах, при отсутствии на линии непрерывного контроля смеси газов, используемых для наполнения.
Должны выполняться испытания на содержание воды, если не установлено иное.
Возможно использование других методик отбора образцов и испытаний при наличии как минимум такого же уровня обеспечения качества.
41. Завершающие испытания переносных криогенных емкостей должны включать испытания на подлинность и количественное определение продукта в каждой емкости, если иное не предусмотрено в регистрационном досье. Выборочный посерийный контроль может использоваться только в случае, если было продемонстрировано, что перед повторным наполнением критические характеристики остаточного газа в каждой емкости остались без изменений.
42. Не требуется проведение отбора образцов после повторного наполнения криогенных емкостей потребителей в месте использования (резервуары в медицинских учреждениях или переносные криогенные емкости) из специально предназначенных цистерн при наличии сопровождающего поставку документа, подтверждающего качество их содержимого. Однако после последовательных повторных наполнений следует удостовериться, что качество газа в емкостях поддерживается на установленном уровне.
43. Не требуется сохранять контрольные и архивные образцы серий продукции, если это не предусмотрено документацией.
44. Не требуется проведение продолжающегося изучения стабильности, если первичное изучение стабильности было заменено библиографическими данными.
Транспортирование газов
45. Наполненные газовые баллоны и переносные криогенные емкости должны быть защищены во время транспортирования, в частности, доставляться заказчикам в чистом состоянии, соответствующем условиям их дальнейшего использования.
Термины и определения
«амбулаторный криогенный сосуд» (home cryogenic vessel) - переносной термически изолированный контейнер, сконструированный для хранения сжиженного кислорода и использования газообразного кислорода на дому у пациента;
«баллон» (cylinder) - контейнер обычно цилиндрической формы, приспособленный для сжатого, сжиженного или растворенного газа и оснащенный приспособлением для регулировки спонтанного вытекания газа при атмосферном давлении и комнатной температуре;
«газ» (gas) - вещество или смесь веществ, которые при давлении 1,013 бар и температуре плюс 200 С находятся полностью в газообразном состоянии или при температуре плюс 500 С давление их паров превышает 3 бар;
«газ как активная фармацевтическая субстанция» (active substance gas) - газ, предназначенный для использования в качестве активной фармацевтической субстанции для производства лекарственного препарата;
«группа (связка) баллонов» (cylinder bundle) - собранные и закрепленные вместе баллоны, подключенные через распределительный коллектор и транспортируемые и используемые как единое целое;
«испытание гидростатическим давлением» (hydrostatic pressure test) - испытание, проводимое с целью обеспечения безопасности в соответствии с требованиями законодательства государств и международных норм для проверки того, что баллоны или резервуары могут выдержать запланированное высокое давление;
«клапан» (valve) - устройство для открывания и закрывания контейнера;
«клапан удержания остаточного давления» (minimum pressure retention valve) - клапан, установленный на баллоне и поддерживающий в использованном баллоне давление выше атмосферного с целью предотвращения контаминации внутреннего объема баллона;
«контейнер» (container) - криогенный сосуд (бак, цистерна или мобильная криогенная емкость другого типа), баллон, связка баллонов или любая другая упаковка, которые находятся в непосредственном контакте с медицинским газом;
«криогенный газ» (cryogenic gas) - газ, который сжижается при давлении 1,013 бар и температуре ниже минус 150 0 С;
«максимальный теоретический остаточный уровень примеси» (maximum theoretical residual impurity) - газообразная примесь от возможного обратного потока газов, оставшаяся после предварительной обработки баллонов перед их наполнением. Расчет максимального теоретического уровня примеси имеет значение только для сжатых газов в предположении, что эти газы ведут себя как идеальные;
«медицинский газ» (medicinal gas) - любой газ или смесь газов, являющиеся лекарственными препаратами;
«обратный клапан» (non-return valve) - клапан, который позволяет потоку проходить только в одном направлении;
«откачивать» (evacuate) - удалять остаточный газ из контейнера (системы) с помощью вакуума до давления меньше чем 1,013 бар;
«переносной криогенный сосуд» (mobile cryogenic vessel) - переносной термически изолированный контейнер, сконструированный для хранения в нем веществ в жидком состоянии, в контексте настоящего документа этот термин не включает в себя понятие «цистерна»;
«продувка» (purge) - удаление остаточного газа из контейнера (системы) путем первоначального нагнетания давления с помощью используемого газа с последующим сбросом давления газа до 1,013 бар;
«разделение воздуха» (air separation) - разделение атмосферного воздуха на составляющие его газы путем фракционной дистилляции при криогенных температурах;
«распределительный коллектор» (manifold) - оборудование или устройство, сконструированное для одновременного опорожнения или наполнения газом одного или более контейнеров;
«резервуар» (tank), «стационарный криогенный сосуд» (fixed cryogenic vessel) - стационарный термически изолированный контейнер для хранения сжиженного или криогенного газа;
«сброс давления» (vent) - операция по удалению остаточного газа из контейнера (системы) до величины остаточного давления 1,013 бар путем соединения контейнера (системы) с атмосферным воздухом;
«сжатый газ» (compressed gas) - газ, который расфасован под давлением с целью транспортирования и остается полностью в газообразном состоянии при температуре выше минус 50 0 С;
«сжиженный газ» (liquefied gas) - газ, который расфасован с целью транспортирования и остается частично жидким или твердым при температуре выше минус 50 0 С;
«цистерна» (tanker) - термически изолированный контейнер, установленный на транспортное средство для перевозки сжиженного или криогенного газа.
ПРИЛОЖЕНИЕ № 7
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству лекарственных растительных препаратов
Принцип
Лекарственные растительные препараты имеют сложную природу и разнообразные характеристики, в связи с чем при их производстве особую роль играет контроль исходных материалов, условий хранения и переработки.
Исходными материалами при производстве лекарственных растительных препаратов могут быть необработанные растения, растительное сырье или промежуточные продукты. Растительное сырье должно иметь требуемое качество, а подтверждающие это данные должны быть предоставлены производителю лекарственного растительного препарата. Для обеспечения постоянного качества растительного сырья может потребоваться более подробная информация о способе его получения (выращивания). Отбор семян, условия культивирования и сбора урожая являются важными аспектами качества растительного сырья и могут влиять на постоянство качества готового лекарственного препарата. Рекомендации в отношении соответствующей системы обеспечения качества по правилам надлежащего выращивания и сбора растений приведены в Руководстве по правилам надлежащего выращивания и сбора исходного сырья растительного происхождения (Guideline on Good Agricultural and Collection Practice for starting materials of herbal origin) Комитета порастительным лекарственным препаратам Европейского агентства по лекарственным средствам (The Committee on Herbal Medicinal Products - HMPC). До принятия в рамках Союза соответствующего акта рекомендуется пользоваться указанным руководством.
Настоящее приложение распространяется на все исходные материалы растительного происхождения: лекарственные растения, растительное сырье и промежуточные продукты из растительного сырья.
Иллюстрация применения различных правил, в том числе Правил надлежащей производственной практики Евразийского экономического союза к производству лекарственных растительных препаратов, приведена в таблице.
|
Виды работ |
Правила надлежащего выращивания и сбора (GACP) |
Часть II Правил надлежащей производственной практики Евразийского экономического союза |
Часть I Правил надлежащей производственной практики Евразийского экономического союза |
|
Культивирование и сбор растений, водорослей, грибов и лишайников, сбор экссудатов (выделений) |
|
|
|
|
Резка и сушка растений, водорослей, грибов, лишайников и экссудатов (выделений) * |
|
|
|
|
Отжим растений и перегонка** |
|
|
|
|
Измельчение, обработка экссудатов, экстракция из растений, фракционирование, очистка, концентрирование или ферментация растительных субстанций |
|
|
|
|
Дальнейшая обработка для получения лекарственной формы, включая упаковку лекарственного препарата |
|
|
|
*Производители должны гарантировать, что данные стадии осуществляются в соответствии с установленными требованиями. Для начальных стадий применимы стандарты надлежащей практики выращивания и сбора для исходных материалов растительного происхождения (GACP). Правила надлежащей производственной практики Евразийского экономического союза применяются к производственным стадиям резки и сушки.
**В отношении стадий отжима растений и перегонки (если необходимо, чтобы эти работы составляли неотъемлемую часть операций заготовки с целью сохранения качества продукции в рамках утвержденных спецификаций) считается приемлемым их проведение в условиях заготовки, если культивирование осуществляется в соответствии с GACP. Такие условия следует рассматривать как исключение и обосновывать в документах регистрационного досье. Для таких работ, осуществляемых в условиях заготовки, необходимо обеспечить соответствующую документацию, контроль и валидацию согласно принципам Правил надлежащей производственной практики Евразийского экономического союза. Надзорные органы могут проводить инспектирование таких работ с целью оценки соответствия Правилам надлежащей производственной практики Евразийского экономического союза.
Помещения и оборудование
Зоны хранения
1. Растительное сырье следует хранить в отдельных зонах. Эти зоны должны быть защищены от проникновения в них насекомых и животных, в особенности грызунов. Должны быть приняты эффективные меры по предотвращению распространения любых таких животных и микроорганизмов, привносимых с растительным сырьем, для предотвращения ферментации или роста плесени, а также перекрестной контаминации. Следует использовать выделенные зоны для карантина поступающего растительного сырья и растительного сырья, разрешенного для использования.
2. Зона хранения должна хорошо вентилироваться. Порядок размещения упаковок не должен препятствовать свободной циркуляции воздуха.
3. Особое внимание следует уделять чистоте и надлежащему обслуживанию зон хранения, в особенности в местах образования пыли.
4. Для хранения исходных материалов и растительных препаратов могут потребоваться особые условия в отношении влажности, температуры и защиты от света. Такие условия следует обеспечивать и контролировать.
Производственная зона
5. При отборе проб, взвешивании, смешивании и других технологических операциях с растительным сырьем и промежуточным продуктом, сопровождающихся пылеобразованием, следует принимать особые меры по поддержанию чистоты, а также по предотвращению перекрестного загрязнения (удаление пыли, выделение специальных помещений и т. п.).
Оборудование
6. Оборудование, фильтрующие материалы и др., используемые в производственном процессе, должны быть совместимы с растворителем, используемым для экстракции, для предотвращения какого-либо выделения или нежелательной абсорбции растительного сырья, которые могут повлиять на продукцию.
Документация
Спецификации на исходное сырье
7. Производители лекарственных растительных препаратов должны убедиться в том, что они используют только те исходные материалы растительного происхождения, которые произведены в соответствии с Правилами надлежащей производственной практики Евразийского экономического союза и регистрационным досье (в соответствии с таблицей настоящего приложения). Следует иметь в наличии исчерпывающую документацию касательно аудитов поставщиков исходных материалов растительного происхождения, проведенных либо самим производителем лекарственного растительного препарата, либо по его поручению. Результаты аудитов в отношении растительного сырья являются основополагающими для качества исходных материалов. Производитель должен убедиться в том, что поставщики растительного сырья (препарата) работают в соответствии с правилами надлежащего выращивания и сбора растений (GACP).
8. Чтобы соответствовать требованиям, установленным в главе 4 части I Правил надлежащей производственной практики Евразийского экономического союза, в спецификации на растительное сырье (препараты) следует включать следующие сведения:
научное название растения в соответствии с бинарной системой (род, вид, подвид (разновидность), а также автор (например, Линней) (при необходимости также следует представить другую соответствующую информацию, такую как название сорта и хемотипическую разновидность);
подробные данные о происхождении растения (страна или регион произрастания либо культивирования, время и способ заготовки, вероятно использованные пестициды, возможное радиоактивное загрязнение и т. д.);
информацию о том, какая часть растения используется; информацию о способе сушки, если используют высушенные растения;
описание растительного сырья, а также данные его макро- и микроскопического исследований;
данные о необходимых испытаниях на подлинность, включая при необходимости испытания на подлинность, для ингредиентов с известной терапевтической активностью или маркеров. Если растительное сырье можно фальсифицировать (подменить), то необходимы специфичные дифференцирующие испытания. Для определения подлинности в распоряжении должен быть аутентичный образец для сравнения;
содержание влаги в растительном сырье, определяемое в соответствии с фармакопейными требованиями;
методики количественного определения компонентов с известной терапевтической активностью или, при необходимости, маркеров; методы, пригодные для определения возможной контаминации пестицидами и пределы приемлемости в соответствии с фармакопейными требованиями, а при отсутствии фармакопейных требований - соответствующий валидированный метод, если не обосновано иное;
методики испытаний по определению грибковой и (или) микробной контаминации, включая афлатоксины, другие микотоксины и инвазию паразитами, а также допустимые пределы, при необходимости;
методики испытаний на наличие токсичных металлов, а также на возможные контаминанты и примеси, при необходимости;
методики испытаний на наличие инородных материалов, при необходимости;
другие виды контроля в соответствии с фармакопейными требованиями.
Любую проведенную обработку для снижения грибковой (микробной) контаминации или другой инвазии следует оформлять документально. Необходимо иметь в распоряжении спецификации и методики, которые должны включать подробные сведения о процессе обработки и испытаниях, а также предельные значения остаточной контаминации.
Технологические инструкции
9. В технологических инструкциях должны быть описаны различные операции, осуществляемые с растительным сырьем (например, очистка, сушка, измельчение и просеивание), а также данные о продолжительности и температуре сушки и методах, используемых для контроля размеров фрагментов кусочков или частиц.
10. Должны быть приняты в форме письменного документа инструкции и записи, которые гарантируют, что каждая тара с растительным сырьем проверена с целью обнаружения какой-либо фальсификации (подмены) или наличия посторонних материалов, таких как фрагменты металла или стекла, остатки животных или их экскременты, камни, песок и др., или признаков гниения.
11. Технологические инструкции должны содержать методы удаления посторонних материалов и соответствующие методики очистки (отбора) материала растительного происхождения перед его хранением в качестве разрешенного растительного сырья или перед началом производства.
12. Инструкции по производству растительных препаратов должны включать подробные сведения о растворителе, продолжительности и температуре экстрагирования, информацию о любых стадиях концентрирования и используемых способах.
Контроль качества
Отбор проб
13. Поскольку растительное сырье по своей природе неоднородно, отбор проб из него должен осуществлять персонал, обладающий специальными навыками. Каждую серию следует идентифицировать по документации на эту серию.
14. Следует сохранять контрольные образцы растительного сырья. При производстве порошков следует сохранять образцы неизмельченного растительного сырья.
15. Персонал, проводящий контроль качества, должен иметь специальную подготовку и опыт работы с растительным сырьем, промежуточными продуктами или лекарственными растительными препаратами для проведения испытаний на подлинность и наличие примесей, выявления в полученном сырье роста грибов, заражения амбарными вредителями, выявления неоднородности сырья и др.
16. Подлинность и качество растительного сырья, промежуточных продуктов и лекарственных растительных препаратов следует определять в соответствии с нормативной документацией.
ПРИЛОЖЕНИЕ № 8
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к отбору проб исходных и упаковочных материалов
Принцип
Отбор проб является важной операцией, при которой отбирается только небольшая часть серии. Достоверные заключения по отношению ко всей серии не могут основываться на испытаниях, проведенных на непредставительных пробах. Таким образом, правильный отбор проб является неотъемлемой частью системы обеспечения качества.
Отбор проб рассматривается в пунктах главы 6 части I Правил надлежащей производственной практики Евразийского экономического союза. Настоящее приложение содержит дополнительные указания по отбору проб исходных и упаковочных материалов.
Персонал
1. Персонал, проводящий отбор проб, должен пройти начальное обучение и в последующем регулярно проходить обучение по дисциплинам, имеющим отношение к правильному отбору проб. Такое обучение должно включать рассмотрение следующих вопросов:
порядок отбора проб;
письменные инструкции по отбору проб;
методы и оборудование, используемые при отборе проб;
риск перекрестной контаминации;
меры предосторожности, которые необходимо принимать в отношении нестабильных и (или) стерильных веществ;
важность принятия во внимание результатов визуальной оценки внешнего вида материалов, упаковок и этикеток;
важность документального оформления любых непредвиденных или необычных обстоятельств.
Исходные материалы
2. Подлинность всей серии исходных материалов, как правило, может быть гарантирована только тогда, когда отдельные пробы были отобраны из всех емкостей и испытание на идентичность было проведено для каждой пробы. Допускается отбирать пробы только из части емкостей, если разработана прошедшая валидацию процедура, гарантирующая, что ни одна емкость с исходными материалами не была неправильно маркирована.
3. При такой валидации следует учитывать, по крайней мере, следующие аспекты:
данные о производителе и поставщике (их тип и текущее состояние), а также их понимание требований Правил надлежащей производственной практики Евразийского экономического союза;
наличие системы обеспечения качества у производителя исходных материалов;
условия производства, при которых исходные материалы производят и контролируют;
характер и свойства исходных материалов и лекарственных препаратов, для производства которых они будет использоваться.
При такой системе процедура, прошедшая валидацию и освобождающая от проведения испытаний подлинности исходных материалов в каждой поступающей емкости, может быть приемлема для: исходных материалов, поступающих от одного производителя или с одного предприятия;
исходных материалов, поступающих непосредственно от производителя или в емкости, опечатанной производителем, при условии, что поставщик имеет безупречную репутацию, и проводятся регулярные аудиты системы обеспечения качества производителя покупателем (производителем лекарственного препарата) или официально аккредитованным органом.
Такая процедура не может удовлетворительно пройти валидацию и использоваться для:
исходных материалов, поставляемых посредниками, когда производитель неизвестен или не подвергается аудиту;
исходных материалов, используемых для производства парентеральных лекарственных препаратов.
4. Качество серии исходных материалов может быть оценено при отборе и проведении испытания представительной пробы. Для этой цели могут быть использованы пробы, отобранные для испытаний подлинности. Количество проб, отобранных для получения представительной пробы, должно быть определено статистически и указано в плане отбора проб. Количество отдельных проб, которые могут быть смешаны для формирования средней пробы, также должно быть определено с учетом вида сырья, сведений о поставщике и однородности средней пробы.
Упаковочные материалы
5. В плане по отбору проб упаковочных материалов должно быть учтено, по крайней мере, следующее: полученное количество; требуемое качество;
характер материала (например, первичные упаковочные материалы и (или) печатные упаковочные материалы); методы производства;
сведения о системе обеспечения качества производителя упаковочных материалов, основанные на результатах проведения аудитов.
Количество отбираемых проб должно быть определено статистически и указано в плане по отбору проб.
ПРИЛОЖЕНИЕ № 9
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству жидких и мягких лекарственных форм
Принцип
Жидкие и мягкие лекарственные формы в особенности подвержены микробной и иной контаминации. Поэтому необходимо принимать специальные меры по предупреждению любой контаминации.
Помещения и оборудование
1. Для защиты от контаминации при производстве и перемещении продукции рекомендуется использовать закрытые системы. Производственные зоны, в которых находятся открытая продукция или открытые чистые упаковки, как правило, следует эффективно вентилировать отфильтрованным воздухом.
2. Конструкция и расположение реакторов, емкостей, трубопроводов и насосов должны предусматривать удобство их очистки и при необходимости санитарной обработки. В частности, в конструкции оборудования должно быть сведено к минимуму наличие недоступных зон или участков, в которых могут скапливаться остатки продукции, создавая среду для размножения микроорганизмов.
3. По возможности не следует использовать аппаратуру из стекла. Как правило, части оборудования, контактирующие с продукцией, должны быть изготовлены из высококачественной нержавеющей стали.
Производство
4. Следует установить и контролировать качество используемой воды в отношении химической и микробиологической чистоты. Во избежание риска размножения микроорганизмов следует надлежащим образом организовать обслуживание систем подготовки воды. После любой химической санитарной обработки систем подготовки воды их необходимо промыть в соответствии с процедурой, прошедшей валидацию, которая гарантирует полное удаление дезинфицирующих средств.
5. Качество сырья, получаемого в емкостях большого объема, следует проверять до их перемещения в емкости для хранения.
6. Следует контролировать передачу сырья по трубопроводам, чтобы гарантировать их поступление в нужное место.
7. В помещениях, где содержатся открытая продукция или открытые чистые упаковки, не допускается нахождение материалов, от которых возможно отделение волокон и других контаминантов (например, картон или деревянные поддоны).
8. Во время фасовки необходимо обеспечить сохранение однородности смесей, суспензий и т. д. Процессы смешивания и фасовки должны пройти валидацию. Особое внимание необходимо уделять обеспечению однородности смеси в начале, после остановок и в конце процесса наполнения.
9. Если готовый продукт упаковывается не сразу, необходимо установить максимально допустимое время до его упаковки, а также соответствующие условия хранения, которые следует строго соблюдать.
ПРИЛОЖЕНИЕ № 10
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству дозированных аэрозольных лекарственных
препаратов под давлением для ингаляций
Принцип
Производство аэрозольных лекарственных препаратов под давлением с дозирующими клапанами, предназначенных для ингаляций, требует особого внимания из-за специфического характера этой лекарственной формы. Его необходимо осуществлять при условиях, сводящих к минимуму контаминацию микроорганизмами и частицами. Очень важно также обеспечить качество деталей клапана, а в случае суспензий - их однородность.
Общие требования
1. Как правило, используются 2 метода производства и наполнения:
а) Система двух стадийного наполнения (наполнение под давлением). Активная фармацевтическая субстанция суспендируется в пропелленте с высокой температурой кипения, доза суспензии подается в контейнер, вставляется и обжимается клапан и через шток клапана вводится пропеллент с низкой температурой кипения для получения готового лекарственного препарата. При этом поддерживается достаточно низкая температура суспензии активной фармацевтической субстанции в пропелленте для снижения потерь за счет испарения;
b) процесс однократного наполнения (холодное наполнение). Активную фармацевтическую субстанцию суспендируется в смеси пропеллентов и суспензия содержится под давлением или при низкой температуре, или одновременно под давлением и при низкой температуре. Затем проводится наполнение упаковки суспензией в 1 прием.
Помещения и оборудование
2. Производство и наполнение следует проводить по возможности в закрытых системах.
3. Зона, в которой продукция или чистые компоненты содержатся открытыми, должна снабжаться отфильтрованным воздухом и соответствовать требованиям к производственной среде по крайней мере класса D. Входить в зону следует через воздушные шлюзы.
Производство и контроль качества
4. Дозирующие клапаны для аэрозолей являются более сложными по сравнению с большинством устройств, используемых в фармацевтической промышленности. Это должно быть учтено в спецификациях на них, а также при отборе проб и при испытаниях. Особое значение имеет проведение аудита системы обеспечения качества у производителя дозирующих клапанов.
5. Все жидкости (например, жидкие или сжиженные под давлением газообразные пропелленты) должны быть профильтрованы для удаления частиц, размер которых больше 0,2 мкм. При наличии возможности желательно проведение дополнительной фильтрации непосредственно перед наполнением.
6. Контейнеры и клапаны необходимо очищать согласно валидированной процедуре, которая соответствует назначению лекарственного препарата и обеспечивает отсутствие любой контаминации (например, контаминация технологическими вспомогательными материалами (например, смазочными) или микробная контаминация). После очистки клапаны следует хранить в чистых закрытых емкостях. Кроме того, должны быть приняты меры предосторожности, предотвращающие контаминацию во время последующих операций, например, при отборе проб. Упаковки должны поступать на линию наполнения в чистом виде или очищаться на линии непосредственно перед наполнением.
7. Необходимо обеспечить однородность суспензии в точке наполнения в ходе всего процесса наполнения.
8. При использовании метода 2-стадийного наполнения для достижения правильного состава необходимо обеспечить точную массу вводимых веществ на обеих стадиях. Поэтому во многих случаях целесообразен 100-процентный контроль массы на каждой стадии.
9. Контроль после наполнения должен подтвердить отсутствие утечек. Проверку на наличие утечек следует проводить так, чтобы не допустить микробной контаминации или остаточной влаги.
ПРИЛОЖЕНИЕ № 11
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к компьютеризированным системам
Принцип
Настоящее Приложение применяется ко всем типам компьютеризированных систем, используемых в рамках деятельности, регулируемой требованиями Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила). Компьютеризированная система представляет собой набор программных и аппаратных компонентов, которые совместно выполняют определенные функции.
Применение компьютеризированной системы должно быть валидировано, информационно-технологическая инфраструктура должна пройти квалификацию.
Если компьютеризированная система заменяет ручное управление, это не должно приводить к снижению качества продукции, технологического контроля или обеспечения качества. Общие риски процесса не должны возрастать.
Общие требования
1. Управление рисками
Управление рисками должно применяться в течение жизненного цикла компьютеризированной системы и учитывать безопасностьпациентов, целостность данных и качество продукции. В рамках системы управления рисками решения по объему валидационных испытаний и проведению контролей целостности данных должны основываться на обоснованной и документально оформленной оценке рисков компьютеризированной системы.
2. Персонал
Следует поддерживать тесное сотрудничество между всем значимым персоналом, вовлеченным в данный процесс (например, с владельцем процесса, владельцем системы, Уполномоченными лицами и техническим (IT) персоналом). Весь персонал должен иметь соответствующую квалификацию, уровень доступа и нести определенную ответственность для выполнения возложенных на него обязанностей.
3. Поставщики и провайдеры услуг
3.1. Если задействованы третьи лица (например, поставщики, провайдеры услуг), например, для поставки, установки, настройки, задания конфигурации, интегрирования, валидации, технического обслуживания (например, через удаленный доступ), модификации или поддержания компьютеризированных систем, связанных с ними услуг или обработки данных, должны иметься надлежаще оформленные договоры между производителем и любыми третьими лицами. В этих договорах должна быть четко установлена ответственность третьих лиц. Аналогичные требования следует предъявлять к подразделениям информационных технологий производителя.
3.2. Компетентность и надежность поставщиков являются ключевыми условиями выбора провайдеров программного продукта или услуг. Необходимость аудита должна быть основана на оценке рисков.
3.3. Документация, прилагаемая к коммерчески выпускаемым готовым для использования программным продуктам, должна быть рассмотрена уполномоченными представителями заказчика на предмет соответствия требованиям пользователя.
3.4. Информация о системе качества и аудитах поставщиков или разработчиков программного обеспечения и установленных компьютеризированных систем должна быть доступна для предоставления инспекторам по их требованию.
Стадия проекта
4. Валидация
4.1. Валидационная документация и отчеты должны охватывать соответствующие стадии жизненного цикла компьютеризированной системы. Производители должны быть способны обосновать свои стандарты, протоколы, критерии приемлемости, процедуры и записи на основе оценки рисков.
4.2. Валидационная документация должна включать в себя записи контроля изменений (если применимо) и отчеты о любых отклонениях, выявленных в ходе процесса валидации.
4.3. Должен быть в наличии текущий перечень (реестр) всех используемых компьютеризированных систем с указанием их функциональности, регулируемой требованиями Правил.
Для критических компьютеризированных систем должны быть в наличии подробное текущее описание физических и логических взаимосвязей, потоков данных и интерфейсов с другими системами или процессами, требуемые ресурсы всего компьютерного оборудования и программного обеспечения, доступные меры безопасности.
4.4. Спецификации требований пользователя должны описывать необходимые функции компьютеризированной системы на основе документально оформленной оценки рисков и влияния с точки зрения соблюдения Правил. Требования пользователя должны прослеживаться на протяжении всего жизненного цикла компьютеризированной системы.
4.5. Заказчику следует предпринять все меры, гарантирующие, что компьютеризированная система разработана в соответствии с надлежащей системой управления качеством. Поставщик должен быть оценен соответствующим образом.
4.6. С целью валидации компьютеризированных систем, изготовленных по индивидуальному заказу или модифицированных в соответствии с требованиями заказчика, следует разработать документированную процедуру оценки качества и эксплуатационных характеристик компьютеризированной системы на всех этапах ее жизненного цикла с оформлением соответствующих отчетов.
4.7. Следует представить доказательства соответствия методов и схем тестирования компьютеризированной системы. В частности, должны быть рассмотрены пределы параметров системы (процесса), границы данных и обработка ошибок. Следует документально оформить оценку соответствия применения автоматизированных средств тестирования и режимов их работы.
4.8. Если данные переводятся в другой формат или систему данных, валидация должна включать проверку неизменности значения и смысла данных в процессе их миграции.
Стадия эксплуатации
5. Данные
Компьютеризированные системы, осуществляющие электронный обмен данных с другими системами, должны включать соответствующие встроенные средства контроля правильного и безопасного ввода и обработки данных с целью минимизации рисков.
6. Контроль точности
Для критических данных, вводимых вручную, следует предусмотреть дополнительный контроль точности ввода данных. Этот контроль может осуществляться вторым оператором или с помощью валидированных электронных средств. Критичность и потенциальные последствия ошибочного или неправильного ввода данных в систему должны охватываться системой управления рисками.
7. Хранение данных
7.1. Данные должны быть защищены от повреждений как физическими, так и электронными мерами. Сохраненные данные должны проверяться на доступность, читаемость и точность. Доступ к данным должен быть обеспечен на протяжении всего периода их хранения.
7.2. Следует выполнять регулярное резервное копирование всех необходимых данных. Сохранность и точность резервных копий, а также возможность восстановления данных должны быть проверены в процессе валидации и периодически контролироваться.
8. Распечатки
8.1. Необходимо иметь возможность получения четких печатных копий данных, хранящихся в электронном виде.
8.2. Для записей, сопровождающих разрешение на выпуск серии, следует предусмотреть возможность получения распечаток, указывающих, изменялись ли какие-либо данные с момента их первоначального ввода.
9. Контрольные следы
На основе оценки рисков следует уделить внимание встраиванию в систему возможности создания записей всех существенных изменений и удалений, связанных с областью действия Правил (система, создающая «контрольные следы»). Причины таких связанных с Правилами изменений или удалений данных должны быть оформлены документально. Контрольные следы должны быть доступными, иметь возможность их преобразования в понятную для пользователей форму, регулярно проверяться.
10. Управление изменениями и конфигурацией
Любые изменения в компьютеризированной системе, включая конфигурацию системы, должны проводиться только контролируемым способом в соответствии с установленной процедурой.
11. Периодическая оценка
Компьютеризированные системы должны периодически оцениваться для подтверждения того, что они остаются в валидированном состоянии и соответствуют требованиям Правил. Такие оценки должны включать, в случае необходимости, оценку текущего диапазона функциональных возможностей, записей отклонений, сбоев, проблем, истории обновлении (upgrades), отчеты об эксплуатации, надежности, защищенности и о валидационном статусе.
12. Защита
12.1. Должны иметься в наличии физические и (или) логические элементы контроля для обеспечения доступа к компьютеризированной системе только уполномоченным на то лицам. Соответствующие способы предотвращения несанкционированного доступа к системе могут включать в себя использование ключей, карточек доступа, персональных кодов с паролями, биометрических данных, ограничения доступа к компьютерному оборудованию и зонам хранения данных.
12.2. Степень защиты зависит от критичности компьютеризированной системы.
12.3. Создание, изменение и аннулирование прав доступа должно быть зарегистрировано.
12.4. Должна быть разработана система управления данными и документами для идентификации операторов, осуществляющих вход, а также для регистрации изменения, подтверждения или удаления данных, включая дату и время.
13. Управление инцидентами
Все инциденты (непредвиденные случаи), включая системные сбои и ошибки данных, должны быть записаны и оценены. Следует установить основную причину критических сбоев и использовать эту информацию в качестве основы корректирующих и предупреждающих действий.
14. Электронная подпись
Электронные записи могут быть подписаны в электронном виде. Электронные подписи должны:
a) в рамках предприятия иметь такое же значение, как рукописные подписи;
b) быть неразрывно связанными с соответствующими записями;
c) включать время и дату, когда они были поставлены.
15. Выпуск серии
Если для регистрации процедуры одобрения и выпуска серии используется компьютеризированная система, она должна предоставлять доступ для выпуска серии только Уполномоченному лицу, а также должна четко идентифицировать и регистрировать сотрудника, который одобрил и выпустил серию в реализацию. Эти действия должны осуществляться с использованием электронной подписи.
16. Непрерывность работы
С целью обеспечения работоспособности компьютеризированных систем, сопровождающих критические процессы, следует принять меры предосторожности для гарантии непрерывности поддержки этих процессов в случае выхода системы из строя (например, с использованием ручной или альтернативной системы). Время, необходимое для введения в действие альтернативных средств, должно учитывать риски и соответствовать конкретной компьютеризированной системе и сопровождаемому рабочему процессу. Эти меры должны быть надлежащим образом оформлены документально и проверены.
17. Архивирование
Данные могут архивироваться. Эти данные должны проверяться на доступность, удобство чтения и целостность. Если в компьютеризированной системе необходимо провести существенные изменения (например, компьютерного оборудования или программного обеспечения), следует обеспечить и проверить возможность восстановления данных.
Определения
«владелец процесса» (process owner) - лицо, ответственное за рабочий процесс;
«владелец системы» (system owner) - лицо, ответственное за работоспособность и обслуживание компьютеризированной системы, а также за защиту находящихся в ней данных;
«жизненный цикл» (lifecycle) - все стадии существования компьютеризированной системы от формирования первоначальных требований до прекращения эксплуатации, включая проектирование, определение технических требований, программирование, тестирование, установку, работу и обслуживание;
«информационно-технологическая инфраструктура» (IT- infrastructure) - компьютерное оборудование и программное обеспечение, такое как сетевое программное обеспечение и операционные системы, которые делают возможным функционирование приложений;
«компьютеризированная система, изготовленная по индивидуальному заказу» (Bespoke (Customized) computerized system) - индивидуально спроектированная компьютеризированная система для обеспечения конкретного рабочего процесса;
«приложение» (application) - программное обеспечение, установленное на определенной платформе (компьютерном оборудовании) и предоставляющее специальные функциональные возможности;
«серийное программное обеспечение» (commercial of the shelf software) - коммерчески доступное программное обеспечение, пригодность которого для использования продемонстрирована большим количеством пользователей;
«третья сторона» (third party) - стороны, которые не находятся в прямом подчинении держателя лицензии на производство лекарственных средств.
ПРИЛОЖЕНИЕ № 12
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к использованию ионизирующего излучения в производстве
лекарственных препаратов
Производитель продукции, для которой ионизирующее облучение является составной частью технологического процесса, должен также руководствоваться актами, регламентирующими использование ионизирующего излучения при производстве лекарственных средств.
Введение
Ионизирующее излучение может использоваться в производственном процессе для различных целей, включая снижение степени бионагрузки и стерилизацию исходного сырья, компонентов упаковки или продукции, а также обработки препаратов крови.
Используется два вида ионизирующего излучения: гамма-излучение от радиоактивного источника и высокоэнергетическое электронное излучение (бета-излучение), полученное с помощью ускорителя.
При гамма-излучении могут быть использованы два различных режима обработки:
порционный режим: продукция размещается в фиксированном положении вокруг источника излучения и не может быть загружена или выгружена, пока открыт источник ионизирующего излучения;
непрерывный режим: автоматизированная система транспортирует продукцию в камеру для облучения мимо открытого источника ионизирующего излучения, перемещает ее с соответствующей скоростью по заданному маршруту, а затем выводит из камеры.
Радиационные установки с ускорителями электронов: продукцию перемещают через непрерывный или пульсирующий пучок электронов высокой энергии (бета-излучение), развертку которого производят в обоих направлениях перпендикулярно к перемещению продукции.
Ответственность
1. Радиационная обработка может осуществляться непосредственно производителем лекарственных средств или по контракту с организацией, имеющей в распоряжении радиационную установку. При этом каждый из них должен иметь соответствующую лицензию на производство и (или) иные разрешения, предусмотренные законодательством.
2. Производитель лекарственных препаратов несет ответственность за качество продукции, в том числе за результаты воздействия ионизирующего излучения. Организация, проводящая радиационную обработку, несет ответственность за то, чтобы каждая упаковка получила дозу, определенную производителем (в том числе упаковка с продукцией, самая удаленная от источника излучения).
3. Требуемая доза с обоснованными предельными значениями должна быть указана в регистрационном досье на лекарственный препарат.
Дозиметрия
4. Дозиметрия - это измерение поглощенной дозы ионизирующего излучения с помощью дозиметров. Понимание принципов работы и правильное использование техники имеют важное значение для валидации, ввода установки в эксплуатацию и контроля процесса.
5. Калибровка каждой партии рабочих дозиметров должна быть прослеживаемой вплоть до национального или международного эталонов. Срок действия калибровки должен быть установлен, обоснован и строго выдержан.
6. Для определения изменения показаний штатных дозиметров после облучения и при их калибровке, как правило, должен использоваться один и тот же прибор. При использовании разных приборов они должны быть калиброваны в абсолютных единицах поглощения.
7. В зависимости от типа используемых дозиметров необходимо учитывать возможные источники погрешностей, включая влажность, изменения температуры, период времени между облучением и измерением, а также мощность поглощенной дозы.
8. Длину волны прибора, используемого для измерения изменений в поглощении дозиметров, и прибор, используемый для измерения плотности потока, следует регулярно проверять путем калибровки через определенные временные интервалы, установленные на основании стабильности, назначения и способа применения прибора.
Валидация процесса
9. Валидация - это действие, доказывающее, что процесс, то есть получение продукцией заданной поглощенной дозы, достигает ожидаемых результатов. Более полно требования к валидации приведены в актах по использованию ионизирующего излучения в производстве лекарственных средств.
10. Валидация должна включать в себя составление карты дозного поля, чтобы установить распределение поглощенной дозы внутри облучаемого контейнера с продукцией при определенной схеме размещения продукции в нем.
11. Технические требования к процессу облучения, как минимум, должны включать в себя:
a) подробные сведения об упаковке продукции;
b) схему(ы) укладки продукции внутри контейнера для облучения. Если в облучаемом контейнере находятся различные виды продукции, особое внимание необходимо уделять тому, чтобы плотная продукция получила полную дозу и не экранировала другую продукцию. Каждый способ укладки в контейнер разных видов продукции должен быть описан в технической документации и должен пройти валидацию;
c) схему расположения контейнеров вокруг источника (порционный режим) или маршрут облучаемых объектов внутри камеры для облучения (непрерывный режим);
d) верхнее и нижнее предельно допустимые значения поглощенной дозы излучения для продукции (и соответствующие методы дозиметрии);
e) верхнее и нижнее предельные значения поглощенной дозы излучения для облучаемого контейнера и соответствующие методы дозиметрии для контроля этой поглощенной дозы;
f) другие параметры процесса, включая мощность поглощенной дозы, максимальное время экспозиции, число экспозиций, количество циклов облучения и пр.
Если облучение проводят по контракту, то в этом контракте должны быть описаны, по крайней мере, подпункты d) и e), регламентирующие технические требования к процессу облучения.
Ввод установки в эксплуатацию
Общие требования
12. Ввод в эксплуатацию - это экспериментально полученное и документально оформленное доказательство того, что радиационная установка при работе в соответствии с техническими требованиями к процессу постоянно будет работать в заранее установленных пределах. Согласно данному Приложению, заранее установленные пределы - это максимальное и минимальное допустимые значения дозы, предназначенной для поглощения облучаемым контейнером. Изменения в работе установки, которые могут привести к выходу значений поглощенной контейнером дозы за эти пределы, ни при каких условиях не должны происходить без ведома оператора.
13. Ввод в эксплуатацию должен включать в себя следующие элементы:
a) планирование;
b) составление карты дозного поля;
c) документальное оформление;
d) определение требований к повторному вводу установки в эксплуатацию.
Источники гамма-излучения
Конструкция
14. Поглощенная доза, полученная определенной частью облучаемого контейнера в любой определенной точке вокруг излучателя, зависит, в первую очередь, от следующих факторов:
a) активности и геометрии источника излучения;
b) расстояния от источника до контейнера;
c) продолжительности облучения, контролируемой таймером или скоростью движения конвейера;
d) состава и плотности материала, включая другую продукцию между источником и определенной частью контейнера.
15. Суммарная поглощенная доза зависит также от маршрута, по которому движутся контейнеры при непрерывном режиме облучения, или от схемы загрузки при порционном режиме облучения, а также от количества циклов облучения.
16. При фиксированном маршруте (при непрерывном облучении) или при фиксированной схеме загрузки (при порционном режиме облучения), а также при постоянной мощности источника и виде продукции основным параметром установки, контролируемым оператором, является скорость конвейера или время, установленное на таймере.
Составление карты дозного поля
17. При составлении карты дозного поля камера для облучения должна быть заполнена контейнерами с муляжами или представительными образцами продукции однородной плотности.
Дозиметры должны быть расположены не менее чем в трех заполненных контейнерах, которые проходят через излучатель. Эти контейнеры должны быть окружены аналогичными контейнерами или муляжами продукции. Если продукция уложена неравномерно, дозиметры должны быть размещены в большем количестве контейнеров.
18. Расположение дозиметров зависит от размеров облучаемого контейнера. Например, для контейнеров размером 1х1х0,5 м, дозиметры могут располагаться в узлах трехмерной решетки с шагом 20 см с учетом внешней поверхности контейнера. Если предполагаемые зоны с максимальной и минимальной дозами известны из предыдущих опытов, то часть дозиметров может быть изъята из зон со средними значениями доз и помещена в зоны с экстремальными значениями дозы с шагом 10 см.
19. В результате этой процедуры должны быть определены минимальная и максимальная дозы, поглощенные продукцией и поверхностью контейнера при заданных параметрах установки, плотности продукции и схеме загрузки.
20. В идеальном случае для определения карты дозного поля следует использовать эталонные дозиметры, поскольку они имеют большую точность. Также допустимо использование обычных дозиметров, но рекомендуется размещать рядом с ними эталонные дозиметры в местах, где предполагаются минимальная и максимальная дозы, и в обычно контролируемом месте в каждом модельном контейнере для облучения. Полученные значения поглощенной дозы будут иметь случайную погрешность, которая может быть определена путем многократных измерений.
21. Измеренная обычным дозиметром минимальная наблюдаемая доза, необходимая для гарантии того, что все облученные контейнеры получили минимальную требуемую дозу, должна быть установлена на основании знания случайной погрешности измерения штатных дозиметров.
22. Во время определения карты дозного поля параметры установки необходимо поддерживать постоянными, контролировать и регистрировать их. Эти записи вместе с результатами дозиметрии и другими полученными записями следует сохранять.
Радиационные установки с ускорителями электронов
Конструкция
23. Поглощенная доза ионизирующего излучения в продукции зависит, в первую очередь, от следующих основных факторов:
a) характеристики пучка, а именно: энергии электронов, среднего потока пучка, ширины развертки и равномерности пучка по ширине развертки;
b) скорости конвейера;
c) состава и плотности продукции;
d) состава, плотности и толщины материала, находящегося между выходным окном и облучаемой частью продукции;
e) расстояния от выходного окна до контейнера.
24. Основными параметрами, контролируемыми оператором, являются характеристики пучка и скорость конвейера.
Составление карты дозного поля
25. При составлении карты дозного поля дозиметры следует располагать между слоями гомогенного поглотителя, моделирующего реальную продукцию, или между слоями реальной представительной продукции однородной плотности так, чтобы не менее 10 измерений были проведены в пределах максимального пробега электронов. Необходимо также соблюдать требования, изложенные в пунктах 18 - 21 настоящего приложения.
26. При определении карты дозного поля параметры радиационной установки необходимо поддерживать постоянными, контролировать и регистрировать их. Эти записи вместе с результатами дозиметрии и другими полученными записями следует сохранять.
Повторный ввод установки в эксплуатацию
27. Процедура повторного ввода в эксплуатацию должна проводиться заново каждый раз, когда происходят изменения процесса или параметров радиационной установки, способные повлиять на распределение поглощенной дозы в облучаемом контейнере (например, при замене стержней облучателя). Объем работ по повторному вводу в эксплуатацию зависит от степени изменений, внесенных в конструкцию облучателя радиационной установки или в конфигурацию загрузки. При наличии сомнений процедуру повторного ввода установки в эксплуатацию следует провести заново.
Помещения
28. Помещения следует проектировать и эксплуатировать таким образом, чтобы облученные контейнеры были отделены от необлученных во избежание перекрестной контаминации. Если материалы обрабатывают в закрытых контейнерах для облучения, то нет необходимости отделять фармацевтические и нефармацевтические материалы друг от друга при условии, что в последующем не будет риска их контаминации.
Любая возможность загрязнения продукции радионуклидами должна быть исключена.
Технологический процесс
29. Контейнеры с продукцией следует загружать в соответствии со схемой загрузки, установленной в процессе валидации.
30. Во время процесса дозу облучения для облучаемых контейнеров необходимо контролировать с использованием прошедших аттестацию дозиметрических методик. Зависимость между этой дозой и дозой, поглощенной продукцией внутри контейнера, должна быть установлена при валидации процесса и вводе радиационной установки в эксплуатацию.
31. Для того, чтобы различать облученные и необлученные контейнеры, необходимо использовать индикаторы ионизирующего излучения. Однако их не следует использовать в качестве единственного средства различения или как единственный показатель удовлетворительных результатов обработки.
32. Одновременную обработку разных видов продукции в одной загрузке в радиационной камере следует проводить только тогда, когда по результатам эксплуатации установки или по другим данным установлено, что поглощенная доза в каждом отдельном контейнере находится в установленных пределах.
33. Если требуемая доза излучения получается более чем за одну экспозицию или более чем за один проход через радиационную камеру, это должно быть согласовано с держателем регистрационного удостоверения; кроме того, эта доза должна быть получена в течение предварительно установленного промежутка времени. Держатель регистрационного удостоверения должен быть уведомлен о незапланированных перерывах во время облучения, если они удлиняют процесс облучения свыше заранее согласованного времени.
34. Облученная продукция всегда должна быть отделена от необлученной. Способы достижения этого включают в себя использование индикаторов радиации (пункт 31 настоящего приложения) и соответствующей планировки помещений (пункт 28настоящего приложения).
Гамма-излучатель
35. При непрерывном режиме облучения дозиметры должны быть расположены таким образом, чтобы под воздействием излучения одновременно находились не менее двух дозиметров в течение всего процесса.
36. При порционном режиме не менее двух дозиметров должны подвергаться воздействию ионизирующего излучения в местах получения минимальной дозы.
37. При непрерывном режиме облучения должна быть предусмотрена индикация требуемого рабочего положения источника. Положение источника и движение конвейера должны быть связаны блокировкой. Скорость движения конвейера необходимо постоянно контролировать и регистрировать.
38. При порционном режиме облучения перемещение источника и время экспозиции для каждой серии продукции должны контролироваться и регистрироваться.
39. Для получения желаемой дозы следует корректировать время облучения и скорость движения конвейера с учетом распада или дозарядки источника излучения. Срок действия параметров установки или скорости конвейера следует фиксировать документально и строго соблюдать.
Радиационные установки с ускорителями электронов
40. В каждый контейнер должен быть помещен дозиметр.
41. Необходимо непрерывно регистрировать среднее значение потока пучка, энергию электронов, ширину развертки и скорость конвейера. Эти параметры, за исключением скорости конвейера, необходимо контролировать в установленных пределах, определенных во время ввода в эксплуатацию, поскольку они подвержены спонтанным изменениям.
Документация
42. Количество поступивших контейнеров и контейнеров, прошедших облучение и вывезенных с предприятия, должно совпадать и соответствовать значениям, указанным в сопроводительной документации. Любые расхождения должны регистрироваться и расследоваться.
43. Оператор радиационной установки должен удостоверять в письменном виде диапазон значений поглощенных доз, полученных каждым контейнером при каждой загрузке или в серии продукции.
44. Технологические записи и записи по контролю для каждой серии продукции, прошедшей облучение, должны проверяться и подписываться специально назначенным лицом и сохраняться. Метод и место хранения должны быть согласованы между организацией, проводившей облучение, и держателем регистрационного удостоверения на лекарственный препарат.
45. Документация, относящаяся к валидации радиационной установки, должна храниться в течение 1 года после истечения срока годности или, по крайней мере, в течение 5 лет после выпуска последней продукции, прошедшей облучение на этой установке, в зависимости от того, какой период дольше.
Микробиологический контроль
46. Ответственность за микробиологический мониторинг несет производитель лекарственных средств. Этот мониторинг может включать мониторинг производственной среды и контроль продукции перед облучением, как установлено в регистрационном досье.
ПРИЛОЖЕНИЕ № 13
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к лекарственным препаратам для клинических исследований
Принцип
Лекарственные препараты для клинических исследований должны быть произведены в соответствии с Правилами надлежащей производственной практики Евразийского экономического союза. Следует принимать во внимание и другие соответствующие руководства в зависимости от стадии разработки лекарственного препарата. Методы работы должны быть гибкими, чтобы обеспечивать возможность внесения изменений по мере расширения знаний о процессе, и отвечать стадии разработки лекарственного препарата.
При клинических исследованиях могут возникнуть дополнительные риски для субъектов исследований по сравнению с пациентами, которые принимают зарегистрированные лекарственные препараты. Применение Правил надлежащей производственной практики Евразийского экономического союза к производству лекарственных препаратов для клинических исследований необходимо для того, чтобы гарантировать субъектам исследования отсутствие рисков, а также чтобы на результаты клинических исследований не повлияли недостаточные безопасность, качество или эффективность, являющиеся следствием ненадлежащего производства. В равной мере эти требования предназначены для обеспечения постоянства от серии к серии одного и того же лекарственного препарата для клинических исследований, используемого в одном или в разных клинических исследованиях, а также для документального оформления и обоснования изменений в процессе разработки такого лекарственного препарата.
Производство лекарственных препаратов для клинических исследований связано с дополнительной сложностью по сравнению с зарегистрированными лекарственными препаратами из-за отсутствия установленных процедур, разных схем клинических исследований и, как следствие, разных требований к упаковкам, необходимости рандомизации и использования «слепого» метода (маскировки), а также из-за большого риска перекрестной контаминации и перепутывания лекарственных препаратов. Кроме того, данные об эффективности и токсичности лекарственного препарата могут быть неполными, может отсутствовать полная валидация процесса или могут использоваться зарегистрированные лекарственные препараты, которые были переупакованы или некоторым образом модифицированы. В связи с этим персонал должен в полной мере понимать Правила надлежащей производственной практики Евразийского экономического союза в отношении лекарственных препаратов для клинических исследований и пройти соответствующее обучение. Должно быть установлено взаимодействие со спонсорами клинических исследований, на которых лежит вся ответственность по вопросам клинических исследований, включая качество лекарственных препаратов для клинических исследований. Возросшая сложность технологических процессов требует применения высокоэффективной системы обеспечения качества.
В настоящем приложении также установлены требования к оформлению заказов, отгрузке, транспортированию и возврату материалов для клинических исследований, которые взаимосвязаны и дополняют правила надлежащей клинической практики.
Лекарственные препараты, не являющиеся
исследуемыми средствами
Субъектам клинических исследований могут быть предоставлены лекарственные препараты, которые не являются испытуемым препаратом, плацебо или препаратом сравнения. Такие лекарственные препараты могут применяться для сопутствующей терапии или для оказания медицинской помощи с целью профилактики, диагностики или лечения, и (или) вследствие необходимости обеспечения адекватного медицинского ухода, предусмотренного для субъектов исследований. Такие средства также допускается применять в соответствии с протоколом исследования, для того чтобы вызвать физиологическую реакцию. Эти лекарственные препараты не являются лекарственными препаратами для клинических исследований и могут быть предоставлены спонсором или исследователем. Спонсор должен гарантировать, что данные лекарственные препараты соответствуют запросу (заявке) на разрешение проведения клинического исследования и имеют требуемое для цели исследования качество. При этом он должен принять во внимание источник лекарственных препаратов, являются ли они зарегистрированными лекарственными препаратами и были ли они переупакованы. Рекомендуется привлекать к этой работе уполномоченное лицо и учитывать его мнение.
Лицензирование производства
и подготовка к применению
Как полное производство, так и отдельные стадии производства лекарственных препаратов для клинических исследований, а также различные процессы их разделения, упаковки или их передача подлежат лицензированию. Однако такое лицензирование не требуется для подготовки к применению лекарственных препаратов перед использованием или упаковкой, если эти процессы осуществляют в медицинских учреждениях фармацевты или другие лица, которые имеют полномочия осуществлять такие процессы, а также если лекарственные препараты для клинических исследований предназначены для использования исключительно в этих учреждениях.
В рамках указанных положений под подготовкой к применению следует понимать простой процесс, такой как:
растворение или диспергирование лекарственного препарата для клинических исследований для ввода лекарственного препарата субъекту испытания, или разведение или смешивание лекарственных препаратов (лекарственного препарата) для клинических исследований с другим веществом (веществами), которое применяют как носитель с целью введения лекарственного препарата.
Смешивание нескольких ингредиентов вместе, включая действующее вещество, с целью получения лекарственного препарата для клинических исследований, не является подготовкой к применению.
Лекарственный препарат для клинических исследований уже должен быть в наличии до процесса подготовки к применению.
Процесс подготовки к применению следует, насколько это возможно, осуществлять непосредственно перед введением.
Такой процесс должен быть указан в заявлении на проведение клинических исследований (досье исследуемого лекарственного препарата) и в протоколе клинического исследования или соответствующем документе, имеющемся на клинической базе.
Определения
«досье на исследуемый препарат» (product specification file) - комплект документов, содержащих всю информацию (или ссылки на соответствующие документы), необходимую для составления подробных инструкций по производству, упаковке, контролю качества, выдаче разрешения на выпуск серии и отгрузке лекарственного препарата для клинических исследований;
«заказ» (order) - задание на производство, упаковку и (или) доставку определенного количества единиц лекарственных препаратов для клинических исследований;
«исследователь» (investigator) - лицо, ответственное за проведение клинического исследования в медицинском учреждении. При проведении клинического исследования группой лиц исследователем является руководитель группы, который может называться ответственным исследователем;
«клиническое исследование» (clinical trial) - исследование, проводимое на людях - субъектах исследования для выявления или подтверждения клинических, фармакологических и (или) других фармакодинамических эффектов лекарственных препаратов для клинических исследований и (или) для выявления побочных реакций на него, и (или) для изучения его абсорбции, распределения, метаболизма и выведения с целью оценки его безопасности и (или) эффективности;
«код рандомизации» (randomization code) - перечень, содержащий описание лечения, назначенного каждому субъекту исследования с учетом рандомизации;
«лекарственный препарат для клинических исследований» (investigational medicinal product) - активная фармацевтическая субстанция в лекарственной форме или плацебо, исследуемые или используемые в качестве препарата сравнения при проведении клинического исследования. К лекарственным препаратам для клинических исследований относятся также уже зарегистрированные лекарственные препараты, если способ их применения или производства (лекарственная форма или упаковка) отличается от зарегистрированного, а также в случае их использования по еще не одобренным показаниям или для получения дополнительной информации об уже зарегистрированной лекарственной форме;
«отгрузка» (shipping) - операции по упаковке для отгрузки и по транспортированию заказанных лекарственных препаратов для клинических исследований;
«препарат сравнения» (comparator product) - исследуемый или зарегистрированный лекарственный препарат (т.е. активный контроль), либо плацебо, используемый как контроль в клиническом исследовании;
«производитель (импортер) лекарственного препарата для клинических исследований» (manufacturer (importer) of investigational medicinal products) - лицо, имеющее лицензию на производство или разрешение на ввоз лекарственного препарата для клинических исследований, выданные в установленном порядке;
«рандомизация» (randomisation) - процесс распределения субъектов исследования по основным и контрольным группам с использованием элемента случайности с целью сведения к минимуму возможности необъективного заключения;
«слепое» исследование («слепой» метод)» (blinding) - процедура клинических исследований, при которой одна или более сторон, участвующих в исследовании, не информированы о проводимом терапевтическом назначении. Простой «слепой» метод означает неосведомленность субъекта исследования, а двойной «слепой» метод - неосведомленность о проводимом терапевтическом назначении (назначениях) субъекта исследования, исследователя, наблюдателей и в некоторых случаях лиц, анализирующих полученные данные. В отношении лекарственного препарата для клинических исследований «слепой» метод исследования означает преднамеренное маскирование идентичности этого лекарственного препарата в соответствии с указаниями спонсора. Раскодирование (снятие маскировки) означает раскрытие информации об идентичности лекарственного препарата;
«спонсор» (sponsor) - физическое лицо, предприятие, учреждение или организация, несущие ответственность за начало клинического исследования, его организацию и (или) финансирование.
Управление качеством
1. Система обеспечения качества, разработанная и проверенная производителем или импортером, должна соответствовать требованиям настоящих правил, относящимся к лекарственным препаратам для клинических исследований, должна быть документально оформлена в виде письменных процедур и должна быть доступна спонсору клинического исследования.
2. Спецификации и технологические инструкции на лекарственные препараты для клинических исследований могут изменяться в процессе их разработки, но при этом необходимо обеспечить их полный контроль и прослеживаемость всех изменений.
Персонал
3. Весь персонал, деятельность которого связана с лекарственными препаратами для исследований, должен пройти соответствующее обучение, связанное со спецификой данного вида продукции.
Даже в случаях, когда количество вовлеченного персонала мало, для производства каждой серии должен быть определен отдельный персонал, отвечающий за производство и контроль качества.
4. Уполномоченное лицо должно обеспечить наличие соответствующих систем, отвечающих требованиям настоящего приложения. Для этого уполномоченное лицо должно иметь хорошую подготовку в области разработки лекарственных препаратов и проведения клинических исследований. Руководство для уполномоченного лица по оценке лекарственных препаратов для клинических исследований приведено в пунктах 38 - 41 настоящего приложения.
Помещения и оборудование
5. При работе с лекарственными препаратами для клинических исследований информация о токсичности, активности и сенсибилизирующих свойствах может быть неполной, в связи с чем следует уделять особое внимание сведению к минимуму рисков перекрестной контаминации. Конструкция оборудования и помещений, методы испытаний и контроля и пределы допустимых концентраций остатков после очистки должны учитывать характер этих рисков. Следует обращать внимание на организацию работы производственными циклами (кампаниями), если это возможно. При выборе моющего средства следует учитывать растворимость лекарственного препарата.
Документация
Спецификации и инструкции
6. Спецификации (на исходное сырье, первичные упаковочные материалы, промежуточные продукты, нерасфасованную и готовую продукцию), регламенты, технологические инструкции и инструкции по упаковке должны быть настолько полными, насколько это позволяет существующий уровень знаний о продукте. По ходу разработки лекарственного препарата их следует периодически оценивать и обновлять (при необходимости). В каждой новой версии должны быть учтены самые последние данные, используемая в настоящее время технология, нормативные и фармакопейные требования, новая версия должна также содержать ссылку на предыдущую версию, чтобы обеспечить прослеживаемость. Любые изменения, которые могут иметь последствия для качества лекарственного препарата, в частности для его стабильности и биоэквивалентности, следует вносить в соответствии с письменной процедурой.
7. Следует документально оформлять причины внесения изменений, должны быть исследованы и документально оформлены последствия изменения в отношении качества лекарственного препарата и любых текущих клинических исследований.
Заказ
8. Заказ должен содержать требование на производство и (или) упаковку определенного числа единиц продукции и (или) ее отгрузку. Заказ производителю дается спонсором или лицом, действующим по его поручению. Заказ должен быть оформлен в письменном виде (но может передаваться и электронным способом) и быть достаточно четким во избежание разночтений. Заказ должен быть официально утвержден и иметь ссылку на досье на лекарственный препарат и на протокол клинических исследований.
Досье на лекарственный препарат
9. Досье на лекарственный препарат должно непрерывно обновляться по мере разработки лекарственного препарата, обеспечивая соответствующую прослеживаемость предыдущих версий. Досье должно включать в себя следующие документы (или содержать на них ссылки):
спецификации и аналитические методики на исходное сырье и упаковочные материалы;
спецификации и аналитические методики на промежуточную, нерасфасованную и готовую продукцию; технологические инструкции; методики контроля в процессе производства; утвержденную копию этикетки;
соответствующие протоколы клинических исследований и коды рандомизации (при необходимости);
соответствующие технические соглашения с заказчиками (при необходимости);
данные о стабильности;
условия хранения и транспортирования.
Приведенный выше перечень не предназначен для установления ограничений и не является исчерпывающим. Он может изменяться в зависимости от лекарственного препарата и стадии его разработки. Содержащаяся в досье информация должна служить основой при оценке готовности для приемки и выдачи разрешения на выпуск конкретной серии уполномоченным лицом, которое должно иметь доступ к этой информации. Если разные стадии процесса производства осуществляют на разных участках, где ответственность несут разные уполномоченные лица, допускается вести отдельные досье с ограниченной информацией, имеющей отношение к деятельности на соответствующих участках.
Регламент и технологические инструкции
10. Каждая производственная операция или операция по отгрузке должна выполняться в соответствии с четкой и достаточно полной письменной инструкцией и сопровождаться оформлением записей. Если операция не является повторяемой, то не обязательно составлять регламент и технологические инструкции. Записи имеют особое значение для подготовки окончательных текстов документов, которые будут использоваться при серийном производстве после получения регистрационного удостоверения.
11. Информацию, содержащуюся в досье на лекарственный препарат, следует использовать при разработке детальных письменных инструкций по производству, упаковке, испытаниям для контроля качества, условиям хранения и транспортирования.
Инструкции по упаковке
12. Лекарственные препараты для клинических исследований, как правило, должны упаковываться индивидуально для каждого субъекта исследований. Количество единиц упаковываемой продукции должно быть определено до начала операций по упаковке с учетом количества единиц, необходимых для проведения контроля качества, и архивных образцов для хранения. После окончания упаковки и маркировки необходимо составить материальный баланс, чтобы гарантировать правильный учет каждого вида продукции для каждой стадии производства.
Записи по производству, контролю
и упаковке серии продукции (досье на серию)
13. Записи по производству, контролю и упаковке серии продукции (досье на серию) должны содержать достаточно подробную информацию для точного прослеживания последовательности операций. Эти записи должны содержать все существенные замечания, обосновывающие использованные процедуры или внесенные изменения, которые расширяют существующие знания о лекарственном препарате и позволяют усовершенствовать производственные операции.
14. Записи по производству, контролю и упаковке серии продукции (досье на серию) должны храниться не менее 5 лет после завершения или официального прекращения последнего клинического исследования, в котором была использована эта серия.
Производство
Упаковочные материалы
15. В спецификациях и методиках контроля качества следует предусматривать специальные меры по предотвращению случайного раскодирования из-за различий внешнего вида разных серий упаковочных материалов.
Технологические операции
16. На стадии разработки лекарственного препарата следует определить критические параметры и виды контроля в процессе производства. Временные параметры и виды контроля в процессе производства могут быть получены из приобретенного опыта, в том числе из предыдущих исследований по разработке. Ключевой персонал должен уделять особое внимание разработке необходимых инструкций и постоянному их совершенствованию с учетом опыта, приобретаемого в процессе производства. Установленные и контролируемые параметры должны быть обоснованы в соответствии с имеющейся в данное время информацией.
17. Не обязательно проводить валидацию технологических процессов производства лекарственных препаратов для исследований в объеме, предусматриваемом для серийного производства, но помещения и оборудование должны быть квалифицированы. Для стерильных лекарственных препаратов валидация процессов стерилизации должна проводиться по тем же стандартам, что и для зарегистрированных лекарственных препаратов. При необходимости, для гарантирования безопасности биотехнологических лекарственных препаратов следует доказать инактивацию (удаление) вирусов и (или) других примесей биологического происхождения в соответствии с научными принципами и методами, изложенными в соответствующих руководствах, действующих в данной области.
18. Валидация асептических процессов представляет особую трудность при малых размерах серий продукции. В этих случаях количество первичных упаковок, наполняемых средами, может быть равно наибольшему размеру серии продукции. По возможности (в том числе для имитации процесса) следует наполнять средами большее число единиц продукции для обеспечения большей достоверности результатов. Наполнение и герметизация являются преимущественно ручными или полуавтоматическими операциями, представляющими риск для стерильности. В связи с этим следует уделить повышенное внимание обучению персонала и проведению валидации методов асептического производства с участием каждого оператора.
Требования к препарату сравнения
19. При изменениях лекарственного препарата объем информации о нем (например, по результатам исследования стабильности, сравнительному тесту кинетики растворения, биодоступности) должен быть достаточным для доказательства того, что эти изменения не окажут существенного влияния на исходные параметры качества этого лекарственного препарата.
20. Срок годности препарата сравнения, указанный на первоначальной упаковке, не может быть таким же для переупакованного в другую упаковку препарата, которая может не обеспечить эквивалентный уровень защиты или может быть несовместимой с препаратом. Поэтому спонсор или лицо, действующее от его имени, должны определить приемлемую дату, до которой следует использовать препарат; при этом следует принять во внимание природу препарата, характеристики контейнера и условия, в которых будет храниться препарат. Новый срок годности должен быть обоснован и не может превышать срок годности, указанный на первоначальной упаковке. Срок годности должен согласовываться с длительностью клинического исследования.
Операции по кодированию («слепой» метод)
21. Если лекарственные препараты кодируют, должны быть системы, обеспечивающие достижение и сохранение кодировки, но при необходимости позволяющие идентифицировать закодированную («слепую») продукцию, в том числе номера серий лекарственного препарата до операции по кодированию. Следует предусмотреть возможность быстрой идентификации лекарственного препарата в экстренных случаях.
Код рандомизации
22. В инструкциях должны быть описаны все процедуры по созданию, защите, распределению, обработке и хранению любого кода рандомизации, использованного для упакованных лекарственных препаратов для клинических исследований, а также методы раскрытия кода. Следует ввести соответствующие записи.
Операции по упаковке
23. При упаковке лекарственного препарата для клинических исследований может оказаться необходимым одновременное обращение продукции различных видов на одной и той же упаковочной линии. Должен быть сведен к минимуму риск перепутывания лекарственных препаратов путем выполнения соответствующих процедур и (или) применения специального оборудования и соответствующего обучения персонала.
24. Операции по упаковке и маркировке лекарственных препаратов для клинических исследований могут быть более сложными и более подверженными ошибкам (которые труднее выявлять), чем при производстве зарегистрированных лекарственных препаратов. Особенно это касается лекарственных препаратов с похожим внешним видом при использовании «слепого» метода. В связи с этим требуется принимать особые меры по предотвращению ошибок в маркировке, например, за счет сведения баланса этикеток, очистки линии, контроля в процессе производства специально обученным персоналом.
25. Упаковка должна гарантировать сохранность лекарственного препарата для клинических исследований в надлежащем состоянии при транспортировании и хранении в промежуточных пунктах назначения. Вторичная (потребительская) упаковка должна быть такой, чтобы было сразу заметно ее вскрытие или любое иное вмешательство во время транспортирования.
Маркировка
26. В таблице 1 суммированы требования, содержащиеся в пунктах 26-30 настоящего приложения. Маркировка должна обеспечивать защиту субъекта исследований, возможность прослеживания и идентификации лекарственного препарата и исследования и способствовать правильному применению лекарственного препарата для клинических исследований. На этикетках должна содержаться следующая информация, если не обосновано ее отсутствие (например, при наличии централизованной электронной системы рандомизированного кодирования):
a) наименование (имя), адрес и номер телефона спонсора, контрактной исследовательской организации или исследователя (основное контактное лицо для информации относительно лекарственного препарата, клинического исследования и для экстренного раскодирования);
b) лекарственная форма, способ введения, количество дозированных единиц, и в случае проведения открытого исследования - наименование (шифр) лекарственного препарата и его дозировка (активность);
c) номер серии и (или) код для идентификации содержимого и операции по упаковке;
d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если это не указано в другом месте;
e) идентификационный номер (лечебный номер) субъекта клинического исследования и при необходимости номер визита;
f) фамилия и инициалы исследователя (если не указано в пунктах а) или d));
g) инструкция по применению (может быть приведена ссылка на листок-вкладыш либо другой пояснительный документ, предназначенный для субъекта клинического исследования или лица, которое вводит лекарственный препарат);
h) надпись «Только для клинических исследований» или аналогичная формулировка;
i) условия хранения;
j) срок использования с указанием месяца и года таким образом, чтобы избежать любой неопределенности (может быть указана дата, до которой необходимо использовать лекарственный препарат, срок годности или дата повторного контроля);
k) надпись «Хранить в недоступном для детей месте», за исключением случаев, когда лекарственный препарат предназначен для использования только в условиях стационара.
27. Адрес и номер телефона основного контактного лица для передачи информации относительно лекарственного препарата, клинического исследования и экстренного раскодирования, могут быть не указаны на этикетке, если субъекту исследования предоставлены инструкция по применению или карточка, на которой указаны эти данные, а также дана инструкция держать их при себе постоянно.
28. Данные должны быть приведены на официальном языке (языках) страны, где будет применяться лекарственный препарат для клинических исследований. Данные, приведенные в пункте 26 настоящего приложения, должны находиться как на первичной (внутренней), так и на вторичной (потребительской) упаковке (кроме случаев, описанных в пунктах 29 и 30 настоящего приложения). Требования к содержанию этикеток на первичной (внутренней) и вторичной (потребительской) упаковках приведены в таблице 1. Также на этикетках может быть приведена информация на других языках.
29. Если лекарственный препарат подготовлен для субъекта исследований или лица, которое вводит лекарственный препарат, в первичном контейнере вместе со вторичной (потребительской) упаковкой, которые следует оставлять вместе, и на вторичной (потребительской) упаковке содержатся данные, приведенные в пункте 26 данного Приложения, на этикетке первичного контейнера (или любого укупоренного дозирующего устройства, содержащего первичный контейнер) должна быть указана следующая информация:
a) наименование (имя) спонсора, контрактной исследовательской организации или исследователя;
b) лекарственная форма, способ введения (можно не указывать для твердых лекарственных форм для применения внутрь), количество дозированных единиц и в случае проведения открытого исследования наименование (шифр) лекарственного препарата и его дозировка (активность);
c) номер серии и (или) код для идентификации содержимого и операции по упаковке;
d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если это не указано в другом месте;
e) идентификационный номер (лечебный номер) субъекта клинического исследования и при необходимости номер визита.
Таблица 1
|
a) наименование (имя), адрес и номер телефона спонсора, контрактной исследовательской организации или исследователя (основное контактное лицо для информации относительно лекарственного препарата, клинического исследования и для экстренного раскодирования); b) лекарственная форма, способ введения, количество дозированных единиц, и в случае проведения открытого исследования - наименование (шифр) лекарственного препарата и его дозировка (активность); c) номер серии и (или) код для идентификации содержимого и указание операции по упаковке; d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если это не указано в другом месте; e) идентификационный номер (лечебный номер) субъекта клинического исследования и при необходимости номер визита; f) фамилия и инициалы исследователя (если не указано в пунктах а) или d)); g) инструкция по применению (может быть приведена ссылка на листок-вкладыш либо другой пояснительный документ, предназначенный для субъекта клинического исследования или лица, которое вводит лекарственный препарат); h) надпись "Только для клинических исследований" или аналогичная формулировка; i) условия хранения; j) срок использования с указанием месяца и года таким образом, чтобы избежать любой неопределенности (может быть указана дата, до которой необходимо использовать лекарственный препарат, срок годности или дата повторного контроля); k) надпись «Хранить в недоступном для детей месте», за исключением случаев, когда лекарственный препарат предназначен для использования только в условиях стационара. |
ОБЩИЙ СЛУЧАЙ
|
|
|
ПЕРВИЧНАЯ (ВНУТРЕННЯЯ)
|
||
|
ПЕРВИЧНАЯ(ВНУТРЕННЯЯ)
|
Примечания.
1Адрес и номер телефона основного контактного лица для получения информации относительно лекарственного препарата, клинического исследования и экстренного раскодирования может не содержаться на этикетке, если субъекту исследования предоставлены инструкция по применению лекарственного препарата или карточка, где указаны эти данные, а также дана инструкция держать их при себе все время (см. пункт 27 данного Приложения).
2Не нужно размещать адрес и номер телефона основного контактного лица для информации относительно лекарственного препарата, клинического исследования и экстренного раскодирования.
3Путь введения можно не указывать в случае твердых лекарственных форм для применения внутрь.
4Можно не указывать лекарственную форму и количество дозированных единиц.
5Если на вторичной (потребительской) упаковке содержится информация, приведенная в пункте 26 настоящего приложения.
30. Если первичной упаковкой является блистер или она имеет малый размер, например ампулы, на которых не могут быть размещены данные, приведенные в пункте 26 настоящего приложения, должна быть предусмотрена вторичная упаковка с этикеткой, содержащей эти данные. Тем не менее, на первичной (внутренней) упаковке должны быть указаны:
a) наименование (имя) спонсора, контрактной исследовательской организации или исследователя;
b) способ введения (можно не указывать для твердых лекарственных форм для применения внутрь), в случае проведения открытого исследования наименование (шифр) лекарственного препарата и его дозировка (активность);
c) номер серии и (или) код для идентификации содержимого и операции по упаковке;
d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если это не указано в другом месте;
e) идентификационный номер (лечебный номер) субъекта клинического исследования и при необходимости номер визита.
31. Для пояснения указанной выше информации могут быть использованы символы или пиктограммы. Может быть представлена дополнительная информация, предостережения и (или) инструкции по обращению с лекарственным препаратом.
32. В случае клинических исследований, когда нет необходимости в отдельных процессах производства или упаковки,
при исследовании используются лекарственные средства, зарегистрированные, произведенные или импортированные согласно законодательству государств - членов или праву Союза,
в исследовании принимают участие пациенты с теми заболеваниями, которые соответствуют показаниям к применению, утвержденным при регистрации,
на оригинальном контейнере так, чтобы не закрыть оригинальную этикетку, дополнительно должны быть приведены следующие данные: название (имя) спонсора, контрактного исследовательской организации или исследователя;
номер (код) исследования, позволяющий идентифицировать медицинское учреждение, исследователя и субъекта исследования.
33. Если необходимо изменить дату, до которой необходимо использовать лекарственный препарат для клинических исследований, следует нанести на упаковку дополнительную этикетку. На дополнительной этикетке должна быть указана новая дата, до которой следует использовать лекарственный препарат, а также повторно указан номер серии. Дополнительную этикетку можно наклеивать поверх старой даты использования, но она не должна закрывать исходный номер серии, что необходимо для контроля качества. Эта операция должна осуществляться на предприятии, имеющем лицензию на производство лекарственных средств. Однако, если обосновано, быть передано на аутсорсинг организации осуществляющей хранение лекарственного препарата для клинических исследований или проведено фармацевтом исследовательского центра или под его контролем, а также другим медицинским работником в соответствии с требованиями законодательства государства - члена Союза. Если это невозможно, операцию может (могут) осуществлять монитор(ы) клинического исследования, который прошел соответствующее обучение. Такую операцию следует осуществлять согласно принципам Правил надлежащей производственной практики Евразийского экономического союза в соответствии со специальными и стандартными операционными процедурами и, если применимо, по контракту; проведение операции должно контролировать второе лицо. Нанесение дополнительной этикетки следует надлежащим образом оформлять документально как в документах клинического исследования, так и в записях, относящихся к серии (досье на серию).
Контроль качества
34. Поскольку процессы могут быть не стандартизованными или не в полной мере валидированными, возрастает значение испытаний для обеспечения гарантии того, что каждая серия продукции соответствует спецификации на нее.
35. Контроль качества необходимо осуществлять в соответствии с досье на лекарственный препарат и согласно информации, предоставленной спонсором уполномоченному органу при заявке на проведение клинического исследования. Следует проводить проверку эффективности кодирования и результаты ее оформлять документально.
36. Образцы лекарственных препаратов для клинических исследований хранятся в двух целях: во-первых, для обеспечения наличия образца для аналитических испытаний и, во-вторых, для обеспечения наличия образца готового лекарственного препарата. Таким образом, образцы можно разделить на две категории:
Контрольный образец (reference sample): образцы серии исходного сырья, упаковочных материалов, лекарственного препарата в первичной упаковке или готового лекарственного препарата, которые хранят для проведения анализа в случае возникновения такой необходимости. Следует сохранять образцы с критических промежуточных стадий (например, после которых предусматривается проведение аналитических исследований и выдача разрешений на выпуск) и образцы промежуточных продуктов, которые поставляются за пределы зоны контроля производителя, если стабильность образцов это позволяет.
Архивный образец (retention sample) - образец в окончательной упаковке, отобранный из серии готовой продукции. Его хранят в целях подтверждения идентичности. Например, в течение срока хранения серии может потребоваться осмотр образца или упаковки, маркировки, инструкции по применению, получение информации о номере серии и сроке годности.
Во многих случаях контрольные и архивные образцы готовой продукции идентичны и являются единицами продукции в окончательной упаковке. В таких случаях контрольные и архивные образцы могут рассматриваться как взаимозаменяемые. Контрольные и архивные образцы лекарственного препарата для клинических исследований, в том числе закодированного лекарственного препарата, должны сохраняться не менее двух лет после окончания или официального прекращения последнего клинического исследования, в котором использовалась данная серия (в зависимости от того, какой из периодов дольше).
Следует уделить внимание хранению архивных образцов до тех пор, пока не будет составлен отчет о проведении клинического исследования, чтобы обеспечить возможность подтверждения идентичности лекарственного препарата, что необходимо при расследованиях непредвиденных случаев или противоречивых результатов таких исследований.
37. Место хранения контрольных и архивных образцов должно быть определено в техническом соглашении между спонсором и производителем(ями), необходимо обеспечить к ним своевременный доступ уполномоченного органа.
Контрольные образцы готового лекарственного препарата должны сохраняться в государстве-члене Союза или в третьей стране, если между государством-членом Союза и третьей страной-экспортером существуют соглашения, которые гарантируют, что производитель лекарственного препарата для клинических исследований придерживается требований правил надлежащего производства, как минимум, эквивалентных Правилам надлежащей производственной практики Евразийского экономического союза. В исключительных случаях контрольные образцы готового лекарственного препарата могут сохраняться у производителя в третьей стране, в таком случае это должно быть обосновано и документально оформлено в виде технического соглашения между спонсором, импортером в государство- член Союза и производителем лекарственного препарата в третьей стране.
Количество контрольных образцов должно быть достаточным для проведения не менее чем двукратного аналитического контроля серии продукции в соответствии с требованиями досье на лекарственный препарат, поданного в уполномоченный орган для получения разрешения на проведение клинических исследований.
Для архивных образцов допускается хранить информацию в отношении окончательно упакованных единиц лекарственных препаратов в виде письменных или электронных записей, если такие записи обеспечивают достаточную информацию. В последнем случае система хранения должна соответствовать требованиям приложения № 11 к Правилам надлежащей производственной практики Евразийского экономического союза.
Выдача разрешения на выпуск серий
38. Не допускается выдача разрешения на выпуск лекарственных препаратов для исследований (пункт 43 настоящего приложения) до тех пор, пока Уполномоченное лицо не удостоверит выполнение установленных требований и требований настоящего приложения (пункт 39 настоящего приложения). Уполномоченное лицо должно учитывать факторы, приведенные в пункте 40настоящего приложения.
39. На выполнение Уполномоченным лицом своих обязанностей в отношении лекарственных препаратов для клинических исследований влияют разные факторы, которые перечислены ниже:
a) лекарственный препарат произведен в государстве-члене Союза, но не зарегистрирован в государстве-члене Союза. При подаче заявления на проведение клинических исследований необходимо засвидетельствовать, что лекарственный препарат для клинических исследований произведен и проверен в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза, досье на лекарственный препарат, а также что имеется соответствующая информация, предоставленная спонсором уполномоченному органу.
b) лекарственный препарат зарегистрирован в государстве- члене Союза, поставляется дистрибьюторы, которая находится в государстве-члене Союза, независимо от того, где производится лекарственный препарат. Обязанности, которые указаны выше, остаются теми же, но объем представленных данных может быть ограничен подтверждением того, что лекарственный препарат соответствует заявлению на проведение клинических исследований и любой последующей обработки с целью кодирования, осуществления специальной упаковки или маркировки для этого исследования. Досье на лекарственный препарат также может быть ограниченным по объему (см. пункт 9 настоящего приложения).
c) лекарственный препарат импортирован непосредственно из третьей страны: необходимо засвидетельствовать, что он произведен и проверен в соответствии с правилами надлежащего производства, как минимум, эквивалентными Правилам надлежащей производственной практики Евразийского экономического союза, досье на лекарственный препарат, а также что имеется соответствующая информация, предоставленная спонсором уполномоченному органу при подаче заявления на проведение клинического исследования. Если лекарственные препараты для клинических исследований ввезены из третьей страны и являются объектом соглашения, принятого между государством-членом Союза и этой страной, например таким, как соглашение о взаимном признании, любое подобное соглашение предусматривает применение требований в отношении этого лекарственного препарата, эквивалентных Правилам надлежащей производственной практики Евразийского экономического союза. При отсутствии соглашения о взаимном признании Уполномоченное лицо на основе информации о системе качества производителя должно установить, что применяются требования, эквивалентные Правилам надлежащей производственной практики Евразийского экономического союза. Эту информацию, как правило, получают путем участия в аудите систем качества производителей. И в первом, и во втором случае
Уполномоченное лицо может выполнить оценку соответствия на основании документации, предоставленной производителем из другой страны (см. пункт 40 настоящего приложения).
d) при ввозе препаратов сравнения, когда невозможно получить гарантию того, что каждая серия продукции была произведена в соответствии с требованиями, эквивалентными Правилам надлежащей производственной практики Евразийского экономического союза, Уполномоченное лицо должно засвидетельствовать, что каждая произведенная серия прошла все необходимые виды контроля и испытаний, необходимые для подтверждения ее качества, в соответствии, а также что имеется соответствующая информация, предоставленная спонсором уполномоченному органу при подаче заявления на проведение клинических исследований.
40. При оценке каждой серии продукции перед выдачей разрешения на выпуск следует рассматривать:
записи, относящиеся к серии (досье на серию), в том числе записи по контролю качества, записи по контролю в процессе производства и записи разрешения на выпуск, свидетельствующие о соответствии досье на лекарственный препарат, заказу, протоколу исследования и коду рандомизации. В эти записи должны быть внесены все отклонения или внесенные в плановом порядке изменения, а также любые дополнительные проверки или испытания. Записи должны быть полными и согласованы персоналом, уполномоченным на это в соответствии с системой качества;
условия производства;
данные об валидации оборудования, процессов и методик;
проверку окончательной упаковки;
результаты любых анализов или испытаний, проведенных после импортирования, если необходимо; отчеты о стабильности;
данные о поставщике и проверке условий хранения и транспортирования;
отчеты об аудитах системы качества производителя;
документы, подтверждающие право производителя на производство лекарственных препаратов для клинических исследований (включая препараты сравнения) на экспорт, выданные уполномоченными органами страны-экспортера;
при необходимости нормативные требования в отношении регистрационной документации, применяемые требования правил надлежащего производства и любые официальные подтверждения выполнения требований правил надлежащего производства;
все другие факторы, которые Уполномоченное лицо считает значимыми для качества серии.
Значимость вышеприведенных факторов зависит от страны, в которой производят лекарственный препарат, предприятия- производителя, статуса регистрации лекарственного препарата (зарегистрирован ли он в государстве-члене Союза или в третьих странах), а также от фазы разработки. Спонсор должен гарантировать, что все факторы, принятые во внимание Уполномоченным лицом, выполняющим оценку серии, соответствуют информации, предоставленной уполномоченному органу при подаче заявления на проведение клинических исследований (пункт 44 настоящего приложения).
41. Если лекарственные препараты для клинических исследований производят и упаковывают на разных участках, за которые несут ответственность разные Уполномоченные лица, необходимо выполнять требования приложения № 16 к Правилам надлежащей производственной практики Евразийского экономического союза.
42. Если согласно законодательству государства - члена Союза и праву Союза упаковка или маркировка осуществляется в исследовательском учреждении фармацевтом, участвующим в проведении клинического исследования, или под его наблюдением, либо другим медицинским работником, то контроль этой деятельности не входит в обязанности Уполномоченного лица. Однако спонсор несет ответственность за гарантию того, что работа надлежащим образом документально оформлена и выполнена в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза. По этому вопросу он должен получить информацию от Уполномоченного лица.
Транспортировка
43. Лекарственные препараты для клинических исследований должны оставаться под контролем спонсора до завершения двухэтапной процедуры выдачи разрешения на выпуск: оценки соответствия Уполномоченным лицом и выдачи спонсором разрешения на выпуск для использования в клинических исследованиях после соблюдения требований законодательства государства - члена Союза и права Союза. Оба этапа должны быть оформлены документально, а записи должны храниться непосредственно у спонсора или у лица, которое действует от его имени. Гармонизированный формат сертификата серии для облегчения перемещения лекарственных препаратов для клинических исследований между государствами-членами Союза, приведен в Дополнении 1 к настоящему приложению. Спонсор должен гарантировать, что вся подробная информация, которая приложена к заявлению на проведение клинического исследования, рассмотрена Уполномоченным лицом и отвечает информации, которая утверждена уполномоченными органами. Должен быть оформлен соответствующий договор о выполнении этого требования. С практической точки зрения, наилучшим способом выполнения этих требований является контроль изменений в досье на лекарственный препарат, что должно быть включено в техническое соглашение между Уполномоченным лицом и спонсором.
44. Транспортирование лекарственных препаратов для клинических исследований следует осуществлять в соответствии с инструкциями, предоставленными в распоряжение спонсором или лицом, действующим от его имени.
45. До поставки лекарственных препаратов для клинических исследований к месту проведения исследований должны быть установлены правила по раскодированию лекарственных препаратов уполномоченным на то персоналом.
46. Следует хранить подробный перечень отгруженной продукции, составленный производителем или импортером. Особое внимание следует уделять точности указания наименования и адреса получателя.
47. Передачу лекарственных препаратов для исследований из одного места проведения исследований в другое следует проводить только в исключительных случаях. Порядок такой передачи должен быть установлен стандартной операционной процедурой. Следует проверить совокупность имеющихся о лекарственном препарате сведений за тот период, когда он находился вне контроля производителя, например, с помощью отчетов о мониторинге клинических исследований или регистрации условий хранения на предыдущем месте проведения исследований. Такая проверка должна учитываться при оценке возможности передачи продукции. К участию в проверке необходимо привлекать Уполномоченное лицо. При необходимости лекарственный препарат следует вернуть производителю или другому имеющему на то право производителю для повторной маркировки и для его оценки Уполномоченным лицом. Следует хранить записи и обеспечивать полное отслеживание подобных передач.
Претензии
48. Выводы по результатам любого расследования, проведенного в связи с поступлением претензии по качеству лекарственного препарата, должны быть обсуждены между производителем или импортером и спонсором (если это не одно и то же лицо). В этом должны участвовать Уполномоченное лицо и лица, ответственные за проведение соответствующего клинического исследования, чтобы оценить возможное влияние претензии на клиническое исследование, разработку лекарственного препарата и субъектов исследований.
Отзывы и возвраты
Отзывы
49. Порядок отзыва лекарственных препаратов для клинических исследований и его документального оформления должен быть согласован между спонсором и производителем или импортером (если это не одно и то же лицо). Исследователь и монитор клинического исследования должны понимать свои обязанности при выполнении отзыва.
50. Спонсор должен гарантировать, что поставщик любого препарата сравнения или других лекарственных препаратов, используемых в клиническом исследовании, имеет систему для извещения спонсора о необходимости отзыва любого поставленного лекарственного препарата.
Возвраты
51. Лекарственные препараты для клинических исследований следует возвращать с соблюдением условий, установленных спонсором и изложенных в утвержденных письменных процедурах.
52. Возвращенные лекарственные препараты для клинических исследований должны быть четко обозначены. Их следует хранить в специально предназначенной контролируемой зоне. Следует сохранять записи по учету возвращенных лекарственных препаратов.
Уничтожение
53. Спонсор несет ответственность за уничтожение неиспользованных и (или) возвращенных лекарственных препаратов для клинических исследований. Не допускается уничтожение лекарственных препаратов для клинических исследований без получения письменного разрешения от спонсора.
54. Для каждого места проведения исследований и каждого периода исследования спонсор или лицо, действующее от его имени, должен фиксировать, составлять баланс и проверять количество лекарственного препарата, которое поставлено, использовано и возвращено. Уничтожение неиспользованных лекарственных препаратов для клинических исследований для данного места проведения исследований или данного периода исследований следует осуществлять только после того, как будет проведено расследование и удовлетворительно объяснены любые несоответствия, а также будет составлен материальный баланс. Документальное оформление операций по уничтожению лекарственного препарата необходимо вести таким образом, чтобы можно было представить отчет обо всех операциях. Записи по уничтожению следует хранить у спонсора.
55. В случае уничтожения лекарственных препаратов для клинических исследований спонсору должен быть представлен акт с указанием даты или другой документ об уничтожении. В этих документах следует четко указать номера серий и (или) номера пациентов (или обеспечить возможность их отслеживания), и количество уничтоженных лекарственных препаратов.
ДОПОЛНЕНИЕ № 1
к приложению № 13
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ПРИМЕРНАЯ ФОРМА
содержания сертификата серии
[БЛАНК ПРОИЗВОДИТЕЛЯ]
Содержание сертификата серии
1. Название (названия) лекарственного(ых) препарата(ов) (идентификатора(ов) лекарственного препарата) в соответствии с заявкой на проведение клинического исследования, в зависимости от того, что применимо.
2. Номер(а) EudraCT (Общеевропейская база данных клинических исследований) и кода протокола спонсора (при наличии).
3. Дозировка:
названия и количество в единице дозы всех активных фармацевтических субстанций для каждого лекарственного препарата для клинических исследований (включая плацебо). Способ предоставления такой информации не должен способствовать раскодированию «слепого» исследования.
4. Лекарственная форма.
5. Размер упаковки (контейнера) и тип (например, флаконы, бутыли, блистеры).
6. Номер партии (серии).
7. Дата окончания срока годности (повторного контроля, срок использования).
8. Наименование производителя (производственной площадки) и его адрес, где находится выдающее сертификат уполномоченное лицо.
9. Номер специального разрешения (лицензии) на производство для производственной площадки, указанной в пункте 8.
10. Комментарии (примечания).
11. Дополнительная информация, которая является существенной по мнению уполномоченного лица.
12. Заявление о сертификации.
13. Делается запись:
«Настоящим я подтверждаю, что эта серия соответствует требованиям (указывается соответствующая форма подтверждения, исходя их нижеперечисленных возможных вариантов поставки лекарственных препаратов):
a) лекарственный препарат произведен на территории государства - члена Евразийского экономического союза (далее - государство-член), но не зарегистрирован в государстве-члене Союза. При подаче заявления на проведение клинических исследований необходимо засвидетельствовать, что лекарственный препарат для клинических исследований произведен и проверен в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила), досье на лекарственный препарат, а также что имеется соответствующая информация, предоставленная спонсором уполномоченному органу;
b) лекарственный препарат зарегистрирован в государстве-члене, поставляется дистрибьютором, который находится в государстве-члене, независимо от того, где производится лекарственный препарат. Обязанности, которые указаны выше, остаются теми же, но объем представленных данных может быть ограничен подтверждением того, что лекарственный препарат соответствует заявлению на проведение клинических исследований и любой последующей обработке с целью кодирования, осуществления специальной упаковки или маркировки для этого исследования. Досье на лекарственный препарат также может быть ограниченным по объему (см. пункт 9 приложения № 13 к Правилам);
с) лекарственный препарат импортирован непосредственно из третьей страны, необходимо засвидетельствовать, что он произведен и проверен в соответствии с требованиями, эквивалентными или не ниже изложенных в Правилах, досье на лекарственный препарат, а также что имеется соответствующая информация, представленная спонсором уполномоченному органу при подаче заявления на проведение клинического исследования. Если лекарственные препараты для клинических исследований ввезены из третьей страны и являются объектом соглашения, принятого между государством-членом и этой страной (например, соглашение о взаимном признании), любое подобное соглашение предусматривает применение требований, эквивалентных Правилам, в отношении этого лекарственного препарата. При отсутствии соглашения о взаимном признании уполномоченное лицо на основе информации о системе качества производителя должно установить, что применяются требования, эквивалентные настоящим Правилам. Эту информацию, как правило, получают путем участия в аудите систем качества производителей. И в первом, и во втором случае уполномоченное лицо может выполнить оценку соответствия на основании документации, представленной производителем из другой страны».
14. Фамилия уполномоченного лица, подписавшего сертификат.
15. Подпись.
16. Дата подписания.
Пояснение
Лекарственные препараты для клинических исследований не могут быть использованы в клиническом исследовании в государствах- членах до окончания двухэтапной процедуры, описанной в пункте 43 приложения № 13 к Правилам. На первом этапе должна быть закончена и оформлена документально сертификация каждой серии уполномоченным лицом производителя или импортера в соответствии с пунктом 13 настоящего документа.
Серию лекарственного препарата для клинических исследований, сопровождаемую подписанным уполномоченным лицом сертификатом серии, не следует подвергать дальнейшим проверкам относительно положений, указанных в пункте 13 настоящего документа при ее перемещении в пределах государств-членов, если законодательством государств-членов не предусмотрено иное. Для облегчения свободного перемещения лекарственных препаратов для клинических исследований между государствами-членами содержание таких сертификатов должно соответствовать приведенному выше гармонизированному формату. Этот формат также может быть применен для сертификации серий, предназначенных для использования в государстве-члене, в котором находится производитель или импортер.
ПРИЛОЖЕНИЕ № 14
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к производству лекарственных препаратов, получаемых из
донорской крови или плазмы
Для целей настоящих Требований используются понятия, которые означают следующее:
«компонент крови» (blood component) - терапевтическая составляющая крови (эритроциты, лейкоциты, тромбоциты и плазма), которая может быть подготовлена различными методами;
«кровь» (blood) - цельная кровь, которая взята у донора и обработана для трансфузии или для дальнейшего производства;
«лекарственные препараты, получаемые из донорской крови или плазмы» (medicinal products derived from human blood or human plasma) - лекарственные препараты на основе компонентов крови, которые произведены промышленным способом на государственных или негосударственных предприятиях;
«обработка» (processing) - любой из этапов получения компонентов крови, который осуществляется после забора крови перед получением компонента крови (например, сепарация и заморозка компонентов крови). В настоящих Требованиях под обработкой дополнительно понимают выполняемые в учреждениях по забору крови операции, которые являются специфическими для плазмы, используемой для фракционирования;
«основное досье плазмы» (Plasma Master File - PMF) - отдельный документ, который не входит в регистрационное досье на лекарственный препарат. В нем содержится вся соответствующая подробная информация в отношении характеристик цельной донорской плазмы, используемой как исходное сырье для производства промежуточных фракций (субфракций), вспомогательных веществ и активных фармацевтических субстанций, которые являются частью плазмы, лекарственных препаратов или медицинских изделий;
«ответственное лицо» (Responsible Person) - специально назначенное лицо в учреждениях по забору (проверке) крови, которое несет ответственность за:
обеспечение того, что кровь или ее компоненты были взяты и проверены в каждой единице независимо от их предназначения, а также за то, что (в случае предназначения для трансфузии) их обработка, хранение и отпуск были произведены в соответствии с законодательством государств - членов Евразийского экономического союза;
предоставление соответствующей информации уполномоченным органам (организациям) государств - членов Евразийского экономического союза в отношении предписаний, разрешений, аккредитации или лицензирования;
выполнение в учреждении по забору (проверке) крови всех требований законодательства государств - членов Евразийского экономического союза.
Ответственное лицо должно отвечать следующим условиям в отношении квалификации:
иметь высшее образование в области медицины или биологии;
иметь стаж работы не менее 2 лет в области забора (проверки) донорской крови и компонентов крови или их обработки, хранения или распределения.
Обязанности ответственного лица, указанные в настоящем пункте, могут быть переданы другим лицам, которые должны иметь соответствующую квалификацию и стаж работы для выполнения этих обязанностей.
Учреждения по забору (проверке) крови должны сообщить в уполномоченный орган (организацию) государств - членов Евразийского экономического союза фамилию (имя, отчество) ответственного лица с обязанностями, указанными в настоящем пункте, а также других лиц, указанных в настоящем пункте, вместе с информацией о конкретных обязанностях, которые на них возложены.
Если ответственное лицо или лица, указанные в настоящем пункте, заменяются на постоянной или временной основе, учреждение по забору (проверке) крови должно незамедлительно уведомить уполномоченные органы (организации) государств - членов Евразийского экономического союза о фамилии (имени, отчестве) нового ответственного лица и дате его назначения;
«плазма для фракционирования» (plasma for fractionation) - жидкая часть донорской крови, которая остается после отделения клеточных компонентов крови, отобранная в контейнер с антикоагулянтом, или которая остается после сепарации с помощью непрерывной фильтрации или центрифугирования крови с антикоагулянтом во время процедуры афереза. Плазма для фракционирования предназначена для производства лекарственных препаратов, получаемых из плазмы, в частности, альбумина, факторов свертывания крови и иммуноглобулина человека;
«препараты крови» (blood products) - лекарственные препараты, полученные из донорской крови или плазмы;
«программа фракционирования по контракту для третьих стран» (third countries contract fractionation program) - фракционирование по контракту на предприятии по фракционированию или производству лекарственных препаратов из донорской плазмы с использованием исходного сырья из третьих стран (государств, не являющихся членами Евразийского экономического союза), при этом произведенная продукция не предназначена для обращения на территории Евразийского экономического союза;
«уполномоченное лицо» (Qualified Person) - лицо, назначенное производителем лекарственных средств, которое осуществляет подтверждение соответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза. Обязанности уполномоченного лица детально описаны в главе 2части I Правил надлежащей производственной практики Евразийского экономического союза и Приложении № 16 к Правилам надлежащей производственной практики Евразийского экономического союза;
«учреждение по забору (проверке) крови» (blood establishment) - учреждение, которое несет ответственность за любой аспект забора и проверки донорской крови или компонентов крови независимо от их дальнейшего предназначения, а также за их обработку, хранение и поставку в случае, когда они предназначены для трансфузии. Этот термин не распространяется на банки крови в больницах, но распространяется на учреждения, в которых проводят плазмаферез;
«фракционирование, предприятие по фракционированию» (fractionation, fractionation plant) - технологический процесс на предприятии (предприятии по фракционированию), во время которого разделяют (очищают) компоненты плазмы с помощью различных физических и химических методов (например, осаждение, хроматография).
1. Область применения
1.1. Положения настоящих Требований распространяются на лекарственные препараты, получаемые из донорской крови или плазмы, фракционированной либо импортированной на территорию Евразийского экономического союза (далее - Союз). Настоящие Требования распространяется также на исходное сырье для таких лекарственных препаратов (например, донорскую плазму) и применимы к стабильным фракциям донорской крови или плазмы (например, альбумина), которые включают в изделия медицинского назначения.
1.2. Настоящие Требования устанавливают специальные требования в отношении производства, хранения и транспортирования донорской плазмы, используемой для фракционирования и производства лекарственных препаратов, получаемых из донорской крови или плазмы.
1.3. Настоящие Требования устанавливают специальные положения для случаев, когда исходное сырье импортируется из третьих стран, а также в случаях программ фракционирования по контракту для третьих стран.
1.4. Настоящие Требования не применяются для компонентов крови, предназначенных для трансфузии.
2. Принцип
2.1. Лекарственные препараты, получаемые из донорской крови или плазмы (а также их активные фармацевтические субстанции, используемые в качестве исходного сырья), должны соответствовать требованиям Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила), а также регистрационному досье на лекарственный препарат. Они рассматриваются как биологические лекарственные препараты и исходное сырье, в которых содержатся такие биологические субстанции, как человеческие клетки или жидкости (включая кровь или плазму). Вследствие биологической природы источников сырья последние имеют определенные характерные особенности (например, исходное сырье может быть контаминировано инфицирующими агентами, в особенности вирусами). Поэтому качество и безопасность таких лекарственных препаратов зависят от контроля исходного сырья и источника его происхождения, а также от дальнейших технологических процедур, включая проверку на инфекционные маркеры, удаление и инактивацию вирусов.
2.2. Все активные фармацевтические субстанции, используемые в качестве исходного сырья для лекарственных препаратов, должны отвечать требованиям Правил (см. пункт 2.1 настоящих Требований).
В отношении забора и проверки исходного сырья, получаемого из донорской крови или плазмы, необходимо придерживаться следующих установленных требований:
забор и проверку следует проводить в соответствии с надлежащей системой качества, соответствующими стандартами и спецификациями;
необходимо выполнять действующие требования в отношении прослеживаемости от донора до реципиента и в отношении уведомлений о серьезных нежелательных реакциях и серьезных нежелательных явлениях;
необходимо руководствоваться фармакопейными требованиями.
2.3. Импортированное из третьих стран исходное сырье для производства лекарственных препаратов, получаемых из донорской крови или плазмы, если эти лекарственные препараты предназначены для применения или распределения в государствах - членах Союза (далее - государства-члены), должно отвечать нормам, эквивалентным действующим в государствах-членах в отношении систем качества учреждений по забору (проверке) крови. Также должны соблюдаться установленные требования по прослеживаемости от донора до реципиента и в отношении уведомлений о серьезных нежелательных реакциях и серьезных нежелательных явлениях, а также должно обеспечиваться соответствие действующим требованиям к крови и ее компонентам.
2.4. При выполнении программ фракционирования по договору с третьими странами исходное сырье, импортированное из стран, не являющихся государствами-членами, должно соответствовать требованиям, действующим в государствах-членах. Работы, проводимые в государствах-членах, должны в полной мере соответствовать Правилам. Следует выполнять требования, действующие в Союзе в отношении систем качества учреждений по забору (проверке) крови. Также должны соблюдаться установленные требования по прослеживаемости от донора до реципиента и в отношении уведомлений о серьезных нежелательных реакциях и нежелательных явлениях, а также должно обеспечиваться соответствие действующим требованиям к крови и ее компонентам.
2.5. Правила распространяются на все стадии после забора и проверки крови (например, обработка (включая разделение), заморозка, хранение и транспортирование к производителю). Как правило, эта деятельность должна находиться в сфере ответственности уполномоченного лица предприятия, которое имеет лицензию на производство лекарственных средств. Если специфические этапы обработки, предназначенной для фракционирования плазмы, проводятся в учреждении по забору (проверке) крови, в нем может быть специально назначено уполномоченное лицо, однако требование к времени его нахождению на производственном участке и ответственности могут быть не пропорциональны времени нахождения на производственном участке и ответственности ответственного лица. Для разрешения этой специфической ситуации и обеспечения того, чтобы обязанности уполномоченного лица, предусмотренные законодательством государства-члена и правом Союза, выполнялись надлежащим образом, предприятие по фракционированию (производитель лекарственных препаратов) должно иметь договор с учреждением по забору (проверке) крови. Договор должен отвечать требованиям, указанным в главе 7 части I Правил, в нем устанавливаются соответствующие обязанности и подробные требования для обеспечения качества. Ответственное лицо учреждения по забору (проверке) крови и уполномоченное лицо предприятия по фракционированию (производителя лекарственных препаратов) должны принимать участие в составлении указанного договора. Для подтверждения того, что учреждение по забору (проверке) крови исполняет условия данного договора, уполномоченное лицо должно обеспечить проведение соответствующих аудитов.
2.6. Специальные требования к документации и другие мероприятия в отношении исходного сырья для получаемых из плазмы лекарственных препаратов указываются в основном досье плазмы.
3. Управление качеством
3.1. Управление качеством должно охватывать все стадии от отбора доноров до поставки готовой продукции. Следует выполнять действующие требования в отношении прослеживаемости на этапе, предваряющем поставку плазмы на предприятие по фракционированию, и на самом этапе поставки, а также в отношении всех стадий, связанных с забором и проверкой донорской крови или плазмы, предназначенных для производства лекарственных препаратов.
3.2. Забор крови или плазмы, которые используются в качестве сырья для производства лекарственных препаратов, следует проводить в учреждениях по забору (проверке) крови, а проверку проводить в лабораториях, которые применяют системы качества, отвечающие установленным требованиям, имеют соответствующее разрешение, выданное уполномоченным органом, и подлежат регулярному инспектированию в соответствии с правом Союза и законодательством государств-членов. При наличии у производителя программ фракционирования по контрактам для третьих стран он обязан уведомить об этом уполномоченный орган.
3.3. В случае импорта плазмы из третьих стран она должна поставляться только утвержденными поставщиками (например, учреждениями по забору (проверке) крови, включая внешние склады). Эти поставщики должны быть указаны в спецификациях на исходное сырье, установленных предприятием по фракционированию (производству), и утверждены уполномоченным органом (например, после инспектирования), а также уполномоченным лицом предприятия по фракционированию в Союзе. В пункте 6.8настоящих Требований описана оценка и выдача разрешения на использование плазмы (плазмы для фракционирования) как исходного сырья.
3.4. Предприятие по фракционированию (производитель готовых лекарственных препаратов) в соответствии с письменными процедурами должно проводить квалификацию поставщиков, включая их аудиты. Следует проводить регулярную повторную квалификацию поставщиков с учетом подхода, основанного на оценке рисков.
3.5. Предприятие по фракционированию (производитель готовых лекарственных препаратов) должен заключить письменные договоры с учреждениями по забору (проверке) крови, которые являются поставщиками.
В каждом таком договоре должны быть указаны как минимум следующие сведения:
определение обязанностей и ответственности;
требования к системе качества и документации;
критерии отбора доноров и проведение испытаний;
требования к разделению крови на компоненты крови и плазму;
заморозка плазмы;
хранение и транспортирование плазмы;
прослеживаемость и информирование после сдачи (забора) крови (в том числе о нежелательных явлениях).
На предприятии по фракционированию (у производителя лекарственных препаратов) должны быть в наличии результаты испытаний всех единиц сырья, поставленных учреждением по забору (проверке) крови. Любая стадия, выполненная по субподряду, должна быть предусмотрена письменным договором.
3.6. Для планирования, оценки и документального оформления всех изменений, которые могут оказать влияние на качество и безопасность продукции или ее прослеживаемость, должна быть установлена надлежащая система контроля. Необходимо оценивать потенциальное влияние предлагаемых изменений. Следует определить необходимость проведения дополнительных испытаний или валидации, особенно на стадиях инактивации и удаления вирусов.
3.7. Для минимизации рисков, связанных с инфицирующими агентами и новыми инфицирующими агентами, должны быть внедрена надлежащая система мер в отношении безопасности. Такая система должна включать в себя оценку рисков для того, чтобы:
определить время выдерживания производственного запаса (время внутреннего карантина) перед обработкой плазмы, чтобы изъять дозы, которые вызывают сомнения (дозы, взятые в течение периода, определенного законодательством государств-членов, прежде чем будет установлено, что дозы, взятые от доноров с высоким риском, должны были исключены из обработки (например, в связи с положительным результатом теста));
учитывать все аспекты, связанные со снижением количества вирусов и (или) сокращением испытаний на инфицирующие агенты или их аналоги;
определить возможности снижения количества вирусов, определить размер серии исходного сырья и иные существенные аспекты процесса производства.
4. Прослеживаемость и мероприятия после забора крови
4.1. Должна быть в наличии система, которая дает возможность прослеживаемости от донора до дозы, забранной в учреждении по забору (проверке) крови, до серии лекарственного препарата, а также в обратном направлении.
4.2. Должна быть определена ответственность за прослеживаемость продукции (отсутствие какого-либо этапа не допускается):
от донора и дозы, взятой в учреждении по забору (проверке) крови, до предприятия по фракционированию (это является обязанностью ответственного лица в учреждении по забору (проверке) крови);
от предприятия по фракционированию до производителя лекарственного препарата и какого-либо субподрядчика, независимо от того, является ли он производителем лекарственного препарата или изделия медицинского назначения (это является обязанностью уполномоченного лица).
4.3. Данные, необходимые для полной прослеживаемости, необходимо хранить не менее 30 лет, если иное не установлено законодательством государств-членов.
4.4. Договоры, указанные в пункте 3.5 настоящих Требований, заключенные между учреждениями по забору (проверке) крови (в том числе контрольными лабораториями) и предприятием по фракционированию (производителем), должны гарантировать, что прослеживаемость и мероприятия после забора крови охватывают всю цепь от забора плазмы до всех производителей, ответственных за выдачу разрешения на выпуск готовой продукции.
4.5. Учреждения по забору (проверке) крови должны уведомлять предприятие по фракционированию (производителя) о каком-либо случае, который может повлиять на качество или безопасность продукции, а также о другой важной информации, полученной после приема донора или выдачи разрешения на выпуск плазмы (например, обратную информацию (информацию, полученную после забора крови)). Если предприятие по фракционированию (производитель) находится на территории государства, не являющегося государством- членом, информацию следует сообщить производителю, находящемуся в государстве-члене, ответственному за выдачу разрешения на выпуск лекарственных препаратов. В указанных случаях такая информация, если она имеет отношение к качеству и безопасности готовой продукции, должна быть доведена до сведения уполномоченного органа, в ведении которого находится предприятие по фракционированию (производитель) лекарственных препаратов.
4.6. В случае если результатом инспектирования уполномоченным органом учреждения по забору (проверке) крови является аннулирование специального разрешения (лицензии, сертификата), необходимо также сделать уведомление, как указано в пункте 4.5 настоящих Требований.
4.7. В стандартных операционных процедурах должно быть описано управление информацией, полученной после забора крови, при этом должны быть учтены лицензионные требования и процедуры информирования уполномоченных органов. Необходимо предусмотреть соответствующие мероприятия после забора крови, которые установлены правом Союза и законодательством государств-членов.
5. Помещения и оборудование
5.1. С целью сведения к минимуму микробной контаминации или внесения постороннего материала в серию плазмы оттаивание и объединение единиц плазмы следует производить в зонах, которые соответствуют требованиям класса чистоты не менее D, установленным в Приложении № 1 к Правилам. Следует использовать специальную одежду, включая маски на лице и перчатки. Иные операции с открытой (неупакованной) продукцией в ходе технологического процесса следует осуществлять в условиях, соответствующих требованиям Приложения № 1 к Правилам надлежащей производственной практики Евразийского экономического союза.
5.2. В соответствии требованиями Приложения № 1 к Правилам надлежащей производственной практики Евразийского экономического союза следует осуществлять регулярный мониторинг производственной среды, особенно во время открывания контейнеров с плазмой, а также во время процессов оттаивания и объединения. Должны быть установлены критерии приемлемости.
5.3. При производстве лекарственных препаратов, получаемых из донорской плазмы, должны использоваться соответствующие методы инактивации или удаления вирусов и приниматься соответствующие меры по предотвращению контаминации обработанной продукции еще не обработанной продукцией. Для стадий технологического процесса, которые проводятся после вирусной инактивации, следует использовать специально предназначенные отдельные помещения и оборудование.
5.4. Для снижения рисков контаминации текущего производства вирусами, которые используются во время валидационных испытаний, валидацию методов снижения количества вирусов не следует проводить с использованием производственных технических средств. Валидацию в указанном случае следует проводить в соответствии с нормативными правовыми актами государств-членов.
6. Производство Исходные материалы
6.1. Исходные материалы должны соответствовать требованиям соответствующих фармакопейных статей, а также удовлетворять условиям, которые содержатся в соответствующем регистрационном досье, в том числе в основном досье плазмы. Эти требования должны быть изложены в письменном договоре (см. пункт 3.5 настоящего приложения) между учреждением по забору (проверке) крови и предприятием по фракционированию (производителем). Их следует контролировать с помощью системы качества.
6.2. Исходные материалы для программ фракционирования по контракту для третьих стран должны соответствовать требованиям, указанным в пункте 2.4 настоящего приложения.
6.3. В зависимости от типа забора (например, забор цельной крови или автоматический аферез) могут потребоваться различные стадии обработки. Все стадии обработки (например, центрифугирование и (или) разделение, отбор проб, маркировка, замораживание) должны быть определены в письменных инструкциях.
6.4. Следует избегать какого-либо перепутывания единиц и образцов, особенно во время маркировки, а также какой-либо контаминации (например, при отрезании сегментов трубок (укупоривании контейнеров)).
6.5. Замораживание является критической стадией выделения протеинов, которые в плазме являются лабильными (например, факторов свертываемости). Поэтому замораживание следует осуществлять с помощью валидированных методов как можно быстрее после забора крови. При этом необходимо придерживаться требований соответствующих фармакопейных статей.
6.6. Условия хранения и транспортирования крови или плазмы к предприятию по фракционированию (производителю) должны быть определены и документально оформлены на всех этапах цепи поставки. Об отклонениях от установленной температуры следует уведомлять предприятие по фракционированию (производителя). Необходимо использовать оборудование, которое прошло квалификацию, и процедуры, которые прошли валидацию.
Оценка (выдача) разрешения на выпуск плазмы для
фракционирования, используемой как исходное сырье
6.7. Разрешение на выпуск плазмы для фракционирования (из карантина) может производиться только посредством систем и процедур, которые обеспечивают качество, необходимое для производства готовой продукции. Плазма может быть поставлена предприятию по фракционированию (производителю) только после документального подтверждения ответственным лицом (в случае забора крови (плазмы) в странах, не являющихся государствами-членами, лицом с эквивалентными обязанностями и квалификацией) соответствия плазмы для фракционирования требованиям и спецификациям, установленным в соответствующих письменных договорах, а также того, что все стадии были проведены в соответствии с Правилами надлежащей производственной практики Евразийского экономического союза.
6.8. Использование контейнеров с плазмой для фракционирования при поступлении на предприятие по фракционированию должно быть разрешено уполномоченным лицом. Уполномоченное лицо должно подтвердить, что плазма соответствует всем требованиям фармакопейных статей, и удовлетворяет условиям соответствующего регистрационного досье, в том числе основного досье плазмы, а в случае использования плазмы для программ фракционирования по контракту для третьих стран - требованиям, указанным в пункте 2.4 настоящего приложения.
Обработка плазмы для фракционирования
6.9. Стадии процесса фракционирования различаются в зависимости от продукции и производителя. Как правило, они включают в себя различные операции фракционирования (очистки), а некоторые из них могут способствовать инактивации и (или) удалению возможной контаминации.
6.10. Следует установить и обеспечить соблюдение требований к процессам объединения, отбора проб из объединенной плазмы, фракционирования (очистки) и инактивации (удаления) вирусов.
6.11. Методы, используемые в процессе вирусной инактивации, следует применять со строгим соблюдением валидированных процедур. Эти методы должны соответствовать методам, которые были использованы при валидации процедур вирусной инактивации. Следует выполнять расследование неудавшихся процедур вирусной инактивации. Соблюдение валидированного технологического процесса является особенно важным в процедурах снижения количества вирусов, поскольку отклонения могут представлять риски для безопасности готовой продукции. Должны быть в наличии процедуры, которые учитывают эти риски.
6.12. Какую-либо повторную обработку или переработку можно производить только после проведения мероприятий по управлению рисками для качества и только на определенных стадиях технологического процесса, что указывается в регистрационном досье соответствующего препарата.
6.13. Должно существовать разделение (различение) лекарственных препаратов и промежуточной продукции, которые прошли процедуру инактивации (удаления) вирусов и которые еще не прошли такую процедуру.
6.14. В зависимости от результата проведенного процесса управления рисками (с учетом возможных отличий в эпидемиологических данных) может быть разрешено производство по принципу производственных циклов в случае, если на 1 предприятии обрабатывают плазму (промежуточную продукцию) различного происхождения, включая необходимые процедуры четкого разделения и наличие установленных валилидированных процедур очистки. Требования для таких мероприятий должны основываться на соответствующих нормативных правовых актах государств-членов. Процесс управления рисками устанавливает необходимость использования специального оборудования в случае программ фракционирования по контракту с третьими странами.
6.15. Для промежуточной продукции, предназначенной для хранения, следует установить срок хранения на основании данных о стабильности.
6.16. Должны быть установлены и документально оформлены требования к хранению и транспортированию промежуточной продукции и готовых лекарственных препаратов на всех этапах цепи поставки. Следует использовать оборудование, которое прошло квалификацию, и процедуры, которые прошли валидацию.
7. Контроль качества
7.1. Требования к испытаниям на вирусы или другие инфицирующие агенты следует устанавливать с учетом новых знаний об инфицирующих агентах и наличия вилидированных методов испытаний.
7.2. Первый однородный пул плазмы (например, после отделения криопреципитата от пула плазмы) следует контролировать с использованием валидированных методов с надлежащей чувствительностью и специфичностью согласно соответствующим фармакопейным требованиям.
8. Выдача разрешения на выпуск промежуточной
и готовой продукции
8.1. Разрешается выпуск только тех серий, которые произведены из пулов плазмы, которые были признаны в результате контроля негативными в отношении вирус-маркеров (антител), а также соответствующих фармакопейным требованиям (включая значения в рамках пределов, ограничивающих содержание вирусов) и утвержденным спецификациям (например, основному досье плазмы).
8.2. Выдача разрешения на выпуск промежуточной продукции, предназначенной для дальнейшей обработки внутри предприятия или поставки на другое предприятие, а также выдача разрешения на выпуск готовых лекарственных препаратов должна осуществляться уполномоченным лицом с соблюдением требований утвержденного регистрационного досье.
8.3. Уполномоченное лицо должно осуществлять выдачу разрешения на выпуск промежуточной или готовой продукции, используемой для программ фракционирования по контракту для третьих стран, на основании нормативов, согласованных с заказчиком, а также в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза. Если такие лекарственные препараты не предназначены для реализации на территории государств-членов Союза, к ним могут не применяться фармакопейные требования установленные в государстве-члене.
9. Хранение образцов пулов плазмы
9.1. Один пул плазмы может быть использован для производства нескольких серий и (или) лекарственных препаратов. Контрольные образцы каждого пула плазмы, а также соответствующие записи следует хранить на менее 1 года после окончания срока хранения полученного из этого пула лекарственного препарата с наибольшим сроком хранения.
10. Удаление отходов
10.1. Следует иметь письменные процедуры безопасного хранения и удаления отходов, одноразовых и отклоненных материалов (например, контаминированных единиц, единиц от инфицированных доноров, а также крови, плазмы, промежуточной продукции или готовых лекарственных препаратов с истекшим сроком годности), что должно оформляться документально.
ПРИЛОЖЕНИЕ № 15
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к квалификации и валидации
Принцип
1. В настоящем приложении описаны принципы квалификации и валидации, применимые к производству лекарственных препаратов. С целью доказательства соответствия параметров критических процессов (оборудования) заданным требованиям производители лекарственных средств должны проводить валидацию процессов и оборудования, используемых при производстве лекарственных средств. Валидация также проводится при существенных изменениях в помещениях, оборудовании и процессах, которые могут оказать влияние на качество продукции.
Для определения состава и объема работ по валидации следует использовать подход, основанный на оценке рисков. В соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза, производители должны определить, какая работа по валидации необходима для подтверждения контроля критических аспектов проводимых ими конкретных операций. Существенные изменения, вносимые в помещение, оборудование и процессы, которые могут повлиять на качество продукции, должны пройти валидацию.
Для определения области проведения и объема валидации следует использовать подход, основанный на оценке рисков.
Планирование валидации
2. Всю деятельность по валидации следует планировать. Ключевые элементы программы валидации следует четко определить и оформить документально в основном плане валидации или эквивалентных документах.
3. Основной план валидации должен быть обобщающим документом, составленным в лаконичной, точной и ясной форме.
4. Основной план валидации должен включать в себя, как минимум, следующее:
a) цель проведения валидации;
b) организационная схема деятельности по валидации;
c) перечень всех помещений, систем, оборудования и процессов, подлежащих валидации;
d) форма документации: форму, которую следует использовать для протоколов и отчетов;
e) планирование и график выполнения работ;
f) контроль изменений;
g) ссылки на существующие документы.
5. В случае выполнения крупных проектов при необходимости составляются отдельные основные планы валидации.
Документация
6. Следует разработать письменный протокол с указаниями, каким образом будет проведена квалификация и валидация. Такой протокол должен быть проверен и утвержден. В протоколе должны быть указаны критические этапы и критерии приемлемости.
7. Должен быть подготовлен отчет с перекрестными ссылками на протокол квалификации и (или) валидации, обобщающий полученные результаты, включающий в себя комментарии относительно любых замеченных отклонений и выводы, включая рекомендуемые изменения, необходимые для устранения отклонений. Изменения, внесенные в план, который приведен в протоколе, следует оформлять документально с соответствующим обоснованием.
8. После успешного завершения квалификации следует оформить официальное письменное разрешение для перехода к следующему этапу квалификации и валидации.
Квалификация
Квалификация проекта
9. Первым элементом проведения валидации новых помещений, систем или оборудования является квалификация проекта.
10. Следует показать и документально оформить соответствие проекта требованиям Правил надлежащей производственной практики Евразийского экономического союза.
Квалификация монтажа
11 Квалификацию монтажа следует проводить для новых или модифицированных помещений, систем и оборудования.
12. Квалификация монтажа должна включать в себя в том числе следующие элементы:
a) проверка монтажа оборудования, трубопроводов, вспомогательных систем и приборов на соответствие действующим техническим чертежам и спецификациям;
b) оценка полноты и сопоставление инструкций поставщика по эксплуатации и работе, а также требований к техническому обслуживанию;
c) оценка требований к калибровке;
d) проверка материалов, использованных в конструкциях.
Квалификация функционирования
13. Квалификация функционирования должна следовать за квалификацией монтажа.
14. Квалификация функционирования должна включать в себя в том числе следующие элементы:
a) испытания, исходя из знаний о процессах, системах и оборудовании;
b) испытания функционирования оборудования при рабочих параметрах, равных верхним и нижним допустимым пределам, то есть в условиях «наихудшего случая».
15. Успешное завершение квалификации функционирования должно способствовать окончательному оформлению инструкций по калибровке, эксплуатации и очистке, проведению обучения операторов, а также установлению требований к профилактическому техническому обслуживанию. Это позволит проводить официальную приемку помещений, систем и оборудования.
Квалификация эксплуатации
16. Квалификация эксплуатации выполняется после успешного завершения квалификации монтажа и квалификации функционирования.
17. Квалификация эксплуатации должна включать в себя в том числе следующие элементы:
а) испытания с использованием реальных исходного сырья и материалов, применяемых в производстве, выбранных заменителей с аналогичными свойствами или моделирующего препарата, разработанные на основании знаний о процессе, а также о технических средствах, системах или оборудовании;
b) испытания при рабочих параметрах, равных верхним и нижним допустимым пределам.
18. Квалификация эксплуатации является отдельным этапом работ, в некоторых случаях допускается проводить ее совместно с квалификацией функционирования при обеспечении целесообразности.
Квалификация установленных (используемых) технических средств,
помещений и оборудования
19. Необходимо иметь данные, обосновывающие и подтверждающие соответствие рабочих критических параметров заданным требованиям. Кроме того, инструкции по калибровке, очистке, профилактическому техническому обслуживанию и эксплуатации, по обучению операторов и ведению отчетов должны быть приняты в форме письменных документов.
Валидация процесса
Общие требования
20. Требования и принципы, предусмотренные настоящим приложением, применимы к производству лекарственных форм. Они распространяются на первоначальную валидацию новых процессов, последующую валидацию измененных процессов и повторную валидацию.
21. Валидация процесса, как правило, должна быть завершена до начала реализации и продажи лекарственного препарата (перспективная валидация). В исключительных случаях, когда такая валидация невозможна, может возникнуть необходимость проведения валидации процессов во время текущего производства (сопутствующая валидация). Процессы, которые уже проводились в течение некоторого времени, также подлежат валидации (ретроспективная валидация).
22. Используемые помещения, системы и оборудование должны быть квалифицированы, а аналитические методики испытаний - валидированы. Персонал, принимающий участие в проведении валидации, должен быть соответствующим образом обучен.
23. Следует проводить периодическую оценку помещений, систем, оборудования и процессов с целью подтверждения их работы в соответствии с заданными требованиями.
Перспективная валидация
24. Перспективная валидация должна включать в себя следующие элементы (но не ограничиваться ими):
a) краткое описание процесса;
b) перечень критических стадий процесса, подлежащих исследованию;
c) перечень используемых помещений (оборудования) (включая измерительное (контрольное, регистрирующее) оборудование) с указанием сведений об их калибровке;
d) спецификации на готовую продукцию при выпуске;
e) при необходимости перечень аналитических методик;
f) предлагаемые точки контроля в процессе производства и критерии приемлемости;
g) при необходимости дополнительные испытания, которые следует провести, вместе с критериями приемлемости и валидацией аналитических методик;
h) план отбора проб;
i) методы регистрации и оценки результатов;
j) функции и обязанности;
k) предполагаемый график выполнения работ.
25. С помощью установленного процесса (используя компоненты, соответствующие спецификациям) можно произвести ряд серий готовой продукции при обычных условиях. Теоретически количество выполненных производственных циклов и сделанных наблюдений должно быть достаточным, чтобы позволить установить обычную степень изменчивости и тенденции, а также получить необходимое количество данных для оценки. Для валидации процесса считается достаточным выполнение 3 последовательных серий (циклов), при которых параметры находятся в заданных пределах.
26. Размер серии при валидации должен быть равным размеру серии при промышленном выпуске продукции.
27. Если предполагается продажа или поставка серий, произведенных при валидации, то условия их производства должны полностью соответствовать регистрационному досье и требованиям Правил надлежащей производственной практики Евразийского экономического союза, включая удовлетворительный результат проведения валидации.
Сопутствующая валидация
28. В исключительных случаях допускается начинать серийное производство до завершения программы валидации.
29. Решение о проведении сопутствующей валидации должно быть обосновано, документально оформлено и утверждено лицами, имеющими на это право.
30. Требования к документации для сопутствующей валидации являются такими же, как и требования, установленные для перспективной валидации.
Ретроспективная валидация
31. Ретроспективная валидация может проводиться только для хорошо отработанных процессов. Проведение ее не допускается, если в состав продукции, технологический процесс или оборудование недавно были внесены изменения.
32. Ретроспективная валидация хорошо отработанных процессов основывается на предшествующих данных. При этом требуются составление специального протокола и отчета и проведение обзора данных предшествующей эксплуатации с выдачей заключения и рекомендаций.
33. Источники данных для такой валидации должны включать в себя (но не ограничиваться ими) записи по производству и упаковке серий продукции, контрольные карты производства, журналы проведения технического обслуживания, данные об изменениях в составе персонала, исследования возможностей процесса, данные о готовой продукции, в том числе карты тенденций, а также результаты изучения ее стабильности при хранении.
34. Серии продукции, отобранные для проведения ретроспективной валидации, должны являться представительной выборкой для всех серий, произведенных в течение рассматриваемого периода, в том числе всех серий, не соответствующих спецификациям. Количество серий продукции должно быть достаточным, чтобы доказать стабильность процесса. При проведении ретроспективной валидации процесса могут понадобиться дополнительные испытания архивных образцов для получения необходимого количества или необходимого вида данных.
35. Для оценки стабильности процесса при проведении ретроспективной валидации следует выполнить анализ данных по 10 - 30 последовательно произведенным сериям, однако при наличии соответствующего обоснования количество исследуемых серий может быть уменьшено.
Валидация очистки
36. Валидацию очистки следует проводить для того, чтобы подтвердить эффективность процедуры очистки. Обоснование выбранных пределов для переносимых остатков продукта, моющих средств, а также микробной контаминации должно основываться на свойствах применяемых материалов. Эти предельные значения должны быть достижимыми на практике и проверяемыми.
37. Для обнаружения остатков или контаминантов следует использовать валидированные аналитические методики. Предел обнаружения для каждой аналитической методики должен быть достаточным для того, чтобы обнаружить установленный допустимый уровень остатка или контаминанта.
38. Необходимо проводить валидацию только процедур очистки поверхностей оборудования, контактирующих с продукцией. Требуется также оценить необходимость валидации деталей оборудования, не контактирующих с продукцией. Следует проводить валидацию длительности интервалов времени между окончанием процесса и очисткой, а также между очисткой и началом следующего процесса. Следует заранее установить методы очистки и интервалы времени между проведением очистки.
39. В отношении процедур очистки, связанных с очень сходными продуктами и сходными процессами, допускается выбрать репрезентативный ряд сходных продуктов и сходных процессов. В указанных случаях можно провести 1 валидационное исследование с использованием подхода «наихудший случай», при котором учтены все критические факторы.
40. Для валидации процедуры очистки достаточно успешное проведение 3 последовательных циклов очистки.
41. Метод «испытывать до тех пор, пока не будет чисто» не заменяет валидацию процедуры очистки.
42. Если удаляемые вещества являются токсичными или опасными, то в порядке исключения вместо них могут использоваться продукты, моделирующие физико-химические свойства таких веществ.
Контроль изменений
43. Необходимо иметь принятые в форме письменных документов процедуры, включающие в себя описание действий, которые следует предпринять, если предполагается изменение исходного сырья, компонентов продукта, технологического оборудования, параметров производственной среды (или участка), способа производства или метода контроля или другое изменение, которое может повлиять на качество продукции или воспроизводимость процесса. Процедуры контроля изменений должны обеспечить получение достаточного количества данных для подтверждения того, что измененный процесс позволяет получать продукцию требуемого качества, соответствующую утвержденным спецификациям.
44. Все изменения, которые могут оказать влияние на качество продукции или воспроизводимость процесса, должны быть официально заявлены, документально оформлены и утверждены. Необходимо оценить возможное влияние изменений в помещениях, системах и оборудовании на продукцию, в том числе провести анализ рисков. Следует определить необходимость и объем повторной квалификации и повторной валидации.
Повторная валидация
45. Следует проводить периодическую оценку помещений, систем, оборудования и процессов, включая проведение процедур очистки, для подтверждения их соответствия заданным требованиям. Если существенные изменения отсутствуют, то вместо повторной валидации составляется отчет, свидетельствующий о соответствии помещений, систем, оборудования и процессов установленным требованиям.
Определения
Понятия, использованные в настоящем приложении, означают следующее:
«анализ рисков» (risk analysis) - метод оценки и описания критических параметров при функционировании оборудования, систем или процесса;
«валидация очистки» (cleaning validation) - документально оформленное подтверждение того, что утвержденная процедура очистки обеспечивает такую чистоту оборудования, которая необходима для производства лекарственных средств;
«валидация процесса» (process validation) - документально оформленное подтверждение того, что процесс, выполняемый в рамках установленных параметров, осуществляется эффективно, воспроизводимо и приводит к производству лекарственного препарата, соответствующего заранее установленным спецификациям и показателям качества;
«квалификация монтажа» (installation qualification, IQ) - документально оформленное подтверждение того, что монтаж помещений, систем и оборудования (установленных или модифицированных) выполнен в соответствии с утвержденным проектом, рекомендациями производителя и (или) требованиями пользователя;
«квалификация проекта» (design qualification, DQ) - документально оформленное подтверждение того, что предложенный проект производственных помещений, оборудования или систем является пригодным для применения по назначению;
«квалификация функционирования» (operational qualification, OQ) - документально оформленное подтверждение того, что помещения, системы и оборудование (установленные или модифицированные) функционируют в соответствии с предъявляемыми требованиями во всех предусмотренных режимах работы;
«квалификация эксплуатации» (performance qualification, PQ) - документально оформленное подтверждение того, что помещения, системы и оборудование при совместном использовании работают эффективно и с воспроизводимыми показателями в соответствии с утвержденными требованиями и характеристиками процесса;
«контроль изменений» (change control) - документально оформленный порядок, в соответствии с которым квалифицированные представители различных специальностей рассматривают предложенные или фактически внесенные изменения, которые могут повлиять на валидированное состояние помещений, оборудования, систем или процессов, целью которого является определение необходимости мероприятий, которые должны обеспечить и документально удостоверить поддержание системы в валидированном состоянии;
«моделирующий препарат» (simulated product) - материал, который по своим физическим и, по возможности, химическим характеристикам (например, вязкости, размерам частиц, pH и др.) близок продукту, в отношении которого проводится валидация. Во многих случаях этими характеристиками может обладать серия препарата-плацебо (продукта, не содержащего активного ингредиента);
«наихудший случай» (worst case) - определенные стандартными операционными процедурами условия или комплекс условий, относящиеся к верхним и нижним предельным значениям рабочих параметров процесса и связанным с ними факторам, которые обусловливают наибольшую вероятность появления сбоя в процессе или дефекта продукта по сравнению с идеальными условиями. Такие условия не обязательно приводят к сбою в процессе или появлению дефекта продукта;
«перспективная валидация» (prospective validation) - валидация, выполняемая до начала серийного производства продукции, предназначенной для реализации;
«повторная валидация» (re-validation) - повторение валидации процесса для обеспечения гарантии того, что изменения в процессе (оборудовании), внесенные в соответствии с процедурой контроля изменений, не ухудшают характеристики процесса и качество продукции;
«ретроспективная валидация» (retrospective validation) - валидация серийного процесса производства реализуемого продукта, основанная на собранных данных о производстве и контроле серий продукции;
«система» (system) - комплекс оборудования, имеющего общее назначение;
«сопутствующая валидация» (concurrent validation) - валидация, выполняемая во время текущего (серийного) производства продукции, предназначенной для реализации.
ПРИЛОЖЕНИЕ № 16
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к подтверждению уполномоченным лицом соответствия
серии продукции с целью ее выпуска
1. Область применения
1.1. Настоящим документом устанавливаются порядок подтверждения соответствия, выполняемого уполномоченным лицом, и требования к выпуску серий зарегистрированных или произведенных для экспорта лекарственных препаратов.
1.2. В настоящем документе также рассматриваются случаи, когда производство серии продукции или проведение анализов разделено на несколько этапов, выполняемых в разных местах или разными производителями, а также когда серию промежуточной или нерасфасованной продукции разделяют на 2 и более серий готовой продукции. Настоящий документ может быть также применен к лекарственным препаратам для клинических исследований, на которые распространяются другие положения актов, составляющих право Евразийского экономического союза, и законодательство государств - членов Евразийского экономического союза и специализированные правила, приведенные в приложении № 13 к Правилам надлежащей производственной практики Евразийского экономического союза (далее - Правила).
1.3 Настоящий документ не описывает все возможные меры, принятие которых допустимо с точки зрения законодательства государств - членов Евразийского экономического союза. В настоящем документе не рассматриваются также разрешения на выпуск серии, выдаваемые уполномоченном органом государства - члена Евразийского экономического союза, которое может иметь специфику для некоторых препаратов крови и иммунобиологических лекарственных препаратов.
1.4. Основные требования к выпуску серии продукции приводятся в регистрационном досье. Положения настоящего документа применяются в области, не противоречащей этим основным требованиям.
2. Принцип
2.1. Уполномоченное лицо должно подтвердить соответствие каждой серии готовой продукции установленным требования до ее выпуска на внутренний рынок или на экспорт.
2.2. Целями регулирования процедуры выпуска серии продукции являются:
гарантия того, что серия была произведена и проверена согласно требованиям лицензии на производство, регистрационного досье и Правил или аналогичных правил надлежащего производства другой страны, признанных эквивалентными указанным Правилам, а также любым другим соответствующим законодательным требованиям перед ее выпуском;
гарантия того, что при необходимости исследования дефекта или отзыва серии уполномоченное лицо, которое выдало разрешение на ее выпуск, и соответствующие записи могут быть легко идентифицированы.
3. Введение
3.1. Производство серии лекарственных препаратов, в том числе контроль качества, разделяется на стадии, которые могут выполняться на разных производственных площадках и разными производителями. Каждая стадия должна выполняться согласно соответствующему регистрационному досье, требованиям Правил, актов, составляющих право Евразийского экономического союза, и законодательства государств - членов Евразийского экономического союза. Этим должно руководствоваться уполномоченное лицо, осуществляющее процедуру подтверждения соответствия серии готовой продукции установленным требованиям перед ее выпуском.
3.2. В условиях промышленного производства, как правило, одно уполномоченное лицо не имеет возможности тщательно изучить каждую стадию производства. Уполномоченное лицо, которое подтверждает соответствие серии готовой продукции, по отдельным вопросам может опираться на заключения других уполномоченных лиц. В таких случаях уполномоченное лицо предварительно должно удостовериться в обоснованности такого доверия исходя из личного опыта или на основании подтверждения другими уполномоченными лицами в рамках признаваемой им системы качества.
3.3. При выполнении отдельных стадий производства в третьей стране аналогичные требования к соответствию производства и проведению контроля качества предъявляются и к участнику производства в этой стране. В этом случае производство лекарственных препаратов также должно осуществляться в соответствии с требованиями регистрационного досье. Производитель должен иметь лицензию на осуществление своей деятельности в соответствии с законом своей страны и выполнять требования Правил или правил надлежащего производства, как минимум, эквивалентных указанным Правилам.
3.4. Некоторым используемым в настоящем документе терминам присвоены конкретные значения, приведенные в разделе «Термины и определения» данного документа.
4. Общие требования
4.1. Различные стадии (этапы) производства, ввоза, контроля и хранения одной и той же серии готовой продукции перед ее выпуском могут осуществляться на разных производственных площадках. Все эти производственные площадки должны иметь одну или раздельные лицензии на производство и осуществлять деятельность под контролем, по крайней мере, одного уполномоченного лица, подтверждающего соответствие этой серии установленным требованиям до ее выпуска. Однако надлежащее производство конкретной серии продукции, независимо от числа задействованных участков, должно находиться под общим надзором уполномоченного лица, подтверждающего соответствие этой серии готовой продукции установленным требованиям перед выпуском.
4.2. Различные серии продукции могут производиться или импортироваться и выпускаться в продажу в разных странах, имеющих соглашение с государствами - членами Евразийского экономического союза о взаимном признании условий производства и реализации. При этом держатель регистрационного удостоверения, а также каждая производственная площадка, имеющая право на выпуск серии продукции, должны иметь в своем распоряжении точное указание адреса площадки, на которой была выпущена конкретная серия продукции, и информацию об уполномоченном лице, ответственном за подтверждение соответствия ее качества установленным требованиям.
4.3. Уполномоченное лицо, подтверждающее соответствие серии готовой продукции установленным требованиям перед выдачей разрешения на выпуск, может основывать свое решение на личном знании всех используемых в производстве помещений и процессов, опыте участвовавшего в производстве персонала и применяемой системы качества, в рамках которой осуществляется производство. Оно может также опираться на заключение со стороны одного или более уполномоченных лиц о соответствии промежуточных стадий производства принятой уполномоченным лицом системе качества.
Такое подтверждение, выдаваемое другими уполномоченными лицами, должно быть оформлено документально и должно ясно определять предмет подтверждения соответствия. Проводимые для выполнения этой задачи систематические мероприятия должны быть указаны в письменном договоре.
4.4. Договор, указанный выше, требуется в тех случаях, когда уполномоченное лицо опирается на заключение другого уполномоченного лица. Этот договор должен в целом соответствовать положениям главы 7 Правил. Уполномоченное лицо, подтверждающее соответствие серии готовой продукции установленным требованиям, должно гарантировать выполнение мероприятий, определенных в указанном договоре. Форма такого договора должна соответствовать взаимоотношениям сторон. Например, оно может представлять собой стандартную операционную процедуру в рамках предприятия или официальный договор между различными предприятиями, даже если они входят в одну и ту же группу компаний.
4.5. Указанный договор должен включать в себя обязательство со стороны поставщика нерасфасованного или промежуточного продукта ставить в известность получателя(ей) продукции обо всех отклонениях, результатах, выходящих за рамки спецификаций, несоответствиях требованиям настоящих Правил, расследованиях, претензиях или других событиях, которые должно принимать во внимание уполномоченное лицо, ответственное за подтверждение соответствия серии готовой продукции всем установленным требованиям.
4.6. Если для документального оформления подтверждения соответствия и выдачи разрешения на выпуск серии продукции используют компьютеризированную систему, следует обратить особое внимание на выполнение требований, приведенных в Приложении 11 настоящих Правил.
4.7. При наличии подтверждения соответствия серии готовой продукции, выданного Уполномоченным лицом, не требуется повторения этой процедуры в странах, имеющих с государствами- членами Союза соглашение о взаимном признании результатов подобной процедуры.
4.8. Вне зависимости от конкретных мероприятий по подтверждению соответствия и выпуску серий должна существовать процедура быстрого выявления и отзыва всей продукции, которая может представлять опасность для потребителей вследствие дефекта серии.
5. Проведение испытаний и выпуск серии продукции, произведенной
в государствах - членах Евразийского экономического союза
5.1. Производство расположено на одной лицензированной производственной площадке.
Если все стадии производства и контроля осуществляются на одной производственной площадке, выполнение отдельных проверок может быть передано другим лицам. Уполномоченное лицо этой производственной площадки, подтверждающее соответствие серии готовой продукции, обычно несет персональную ответственность за это в рамках установленной системы качества. Однако оно может принимать во внимание также подтверждение в отношении промежуточных стадий, выданное на участке другими уполномоченными лицами, которые несут ответственность за эти стадии.
5.2. Различные стадии производства выполняются в разных местах.
Если различные стадии производства серии продукции осуществляются на разных производственных площадках в пределах одной организации (независимо от того, распространяется ли на них одна и та же лицензия на производство или нет), то уполномоченное лицо должно отвечать за каждую стадию производства. Подтверждение соответствия серии готовой продукции установленным требованиям должно выполняться уполномоченным лицом производителя, которое либо несет персональную ответственность за все стадии производства, либо принимает во внимание заключения о предшествующих стадиях, сделанные уполномоченными лицами, ответственными за эти стадии.
5.3. Некоторые промежуточные стадии производства осуществляются по контракту другой организацией.
Одна или более промежуточных стадий производства и контроля качества могут выполняться в другой организации по контракту держателем лицензии на производство. Уполномоченное лицо заказчика может принимать во внимание заключение уполномоченного лица исполнителя о соответствующей стадии, но оно несет ответственность за гарантию того, что эта работа выполняется в соответствии с условиями письменного договора. Подтверждение соответствия серии готовой продукции установленным требованиям должно быть выполнено уполномоченным лицом производителя, ответственного за выпуск серии продукции.
5.4. Из серии нерасфасованной продукции на разных производственных площадках производятся несколько серий готовой продукции, которые выпускаются на рынок на основании одного регистрационного удостоверения. Это происходит, например, в случае одного регистрационного удостоверения, если все участки по фасовке находятся на территории одного государства-члена Союза.
5.4.1. Уполномоченное лицо держателя лицензии на производство лекарственных средств, выпускающее серию нерасфасованной продукции, может подтверждать соответствие всех серий готовой продукции перед их выпуском. В этом случае уполномоченное лицо либо берет на себя персональную ответственность за все стадии производства, либо принимает во внимание заключения о качестве серий продукции, полученные от Уполномоченных лиц с мест выпуска серий готовой продукции.
5.4.2. Допускается также подтверждение соответствия каждой серии готовой продукции до момента ее выпуска уполномоченным лицом производителя, который выполнил последнюю производственную операцию, предшествующую выпуску серии готовой продукции. В этом случае оно либо берет на себя персональную ответственность за все стадии производства, либо принимает во внимание заключение о качестве серии, полученное от уполномоченного лица с места выпуска серии нерасфасованной продукции.
5.4.3. Во всех случаях организации производства готовой продукции в разных местах на разных производственных площадках в рамках одного регистрационного удостоверения должно быть определено одно лицо (как правило, уполномоченное лицо производителя серии нерасфасованной продукции), которое несет полную ответственность за выпуск всех серий готовой продукции, полученных из одной серии нерасфасованной продукции. Это лицо должно знать о любых проблемах, связанных с качеством любой серии готовой продукции, и координировать осуществление всех необходимых мер, предпринимаемых в связи с наличием проблемы в отношении серии нерасфасованной продукции.
Хотя номера серий нерасфасованной и готовой продукции необязательно должны совпадать, необходимо документально оформить связь между номерами этих серий, чтобы можно было обеспечить прослеживание при аудите.
5.5. Из серии нерасфасованной продукции в разных местах на разных производственных площадках производится несколько серий готовой продукции, которые выпускаются на основании разных регистрационных удостоверений. Это происходит, например, когда транснациональная компания владеет национальными регистрационными удостоверениями на лекарственный препарат в нескольких государствах-членах Союза или производитель воспроизведенных лекарственных препаратов приобретает нерасфасованный лекарственный препарат и выпускает готовый лекарственный препарат по своему собственному регистрационному удостоверению.
5.5.1. Уполномоченное лицо производителя готовой продукции, подтверждающее соответствие серии готовой продукции установленным требованиям, может либо взять на себя персональную ответственность за все стадии производства, либо основываться на заключении, полученном от уполномоченного лица производителя нерасфасованной продукции.
5.5.2. Любая проблема, связанная с качеством любой серии готовой продукции, источником которой могла явиться исходная серия нерасфасованной продукции, должна быть сообщена уполномоченному лицу, ответственному за подтверждение качества этой серии нерасфасованной продукции. После этого указанное уполномоченное лицо должно предпринять все необходимые действия в отношении всех серий готовой продукции, произведенных из данной серии нерасфасованной продукции. Порядок действий в этом случае должен быть установлен в письменном соглашении.
5.6. Серию готовой продукции закупает и реализует держатель лицензии на производство лекарственных средств в соответствии с собственным регистрационным удостоверением. Это происходит, например, когда предприятие, поставляющее воспроизведенный лекарственный препарат, является держателем регистрационного удостоверения на лекарственный препарат, производимый другим предприятием. В этом случае первое предприятие закупает готовую продукцию, соответствие которой еще не было подтверждено производителем, и выпускает ее на основании собственной лицензии на производство и собственного регистрационного удостоверения.
В этой ситуации уполномоченное лицо предприятия, закупающего продукцию и не имеющего документальных результатов подтверждения ее соответствия, должно само подтвердить соответствие установленным требованиям этой серии готовой продукции перед ее выпуском. При этом уполномоченное лицо закупающего предприятия принимает на себя ответственность за все стадии производства или основывается на заключении о качестве серии продукции уполномоченного лица предприятия-поставщика.
5.7. Лаборатория контроля качества лекарственных препаратов и производитель лекарственных препаратов являются разными организациями.
Уполномоченное лицо, подтверждающее соответствие серии готовой продукции установленным требованиям, может принять на себя ответственность за лабораторные испытания, проведенные подобной лабораторией или учитывать подтверждение в отношении испытаний, выданное другим уполномоченным лицом. При отсутствии такого подтверждения уполномоченное лицо должно знать работу данной лаборатории и методики, применяемые в ней для подтверждения соответствия качества данной готовой продукции.
6. Обязанности уполномоченного лица
6.1. Перед осуществлением подтверждения соответствия серии готовой продукции до ее выпуска уполномоченное лицо должно гарантировать, как минимум, выполнении следующих требований:
a) серия готовой продукции и процесс ее производства соответствуют положениям регистрационного досье;
b) серия готовой продукции произведена в соответствии с требованиями Правил, а для серии продукции, импортируемой из третьих стран - в соответствии с правилами надлежащего производства, по крайней мере, эквивалентными требованиям Правил;
c) основные процессы производства и методы контроля валидированы; учтены фактические условия производства в записях, относящиеся к серии (досье на серию) продукции;
d) любые отклонения или запланированные изменения в технологическом процессе или контроле качества были утверждены ответственными лицами в соответствии с определенной системой. О любых изменениях, требующих внесения изменения в регистрационное досье или лицензию на производство, осведомлен соответствующий уполномоченный орган и получено его разрешение на внесение такого изменения;
e) проведены все необходимые проверки и испытания (в том числе дополнительный отбор проб, инспектирование, проверки и испытания, проведенные ввиду отклонений в технологическом процессе или ввиду плановых изменений);
f) документация по производственному процессу и контролю качества составлена и утверждена уполномоченным персоналом;
g) все аудиты проведены в соответствии с требованиями системы обеспечения качества;
h) приняты во внимание все факторы, которые, по мнению уполномоченного лица, являются существенными для качества данной серии продукции.
Формирование и ведение реестра уполномоченных лиц производителей лекарственных средств Союза осуществляется Евразийской экономической комиссией в соответствии с порядком, утверждаемым Евразийской экономической комиссией.
Уполномоченное лицо может иметь дополнительные обязанности в соответствии с законодательством или должностными инструкциями.
6.2. Уполномоченное лицо, подтверждающее соответствие промежуточной стадии производства согласно пункту 4.3настоящего документа, имеет такие же вышеуказанные обязанности по отношению к этой стадии (если в договоре между уполномоченными лицами не указано другое).
6.3. Уполномоченное лицо должно поддерживать свою квалификацию на современном уровне в свете достижений научно-технического прогресса и учитывать изменения в системе управления качеством, имеющие отношение к продукции, соответствие которой установленным требованиям подтверждает уполномоченное лицо.
6.4. При привлечении уполномоченного лица к подтверждению соответствия серии продукции, которую он знает недостаточно (например, при освоении нового вида продукции или при переходе на другое предприятие), Уполномоченное лицо должно получить соответствующие знания и опыт, необходимые для выполнения этих обязанностей.
В соответствии с национальными требованиями на уполномоченное лицо может быть возложена обязанность уведомлять уполномоченные органы о таком изменении; это может потребовать повторной аттестации.
7. Термины и определения
Определенные слова и словосочетания в настоящем документе используются в определенных значениях, указанных ниже. Также следует обращаться к общему разделу «Термины и определения» Правил.
«импортер» (importer) - держатель лицензии на импорт лекарственных препаратов из третьих стран, если таковая предусмотрена законодательством;
«оценка соответствия серии готовой продукции» (certification of the finished product batch) - документальное оформление соответствия серии готовой продукции установленным требованиям до выпуска серии;
«подтверждение» (confirmation) - подписанное свидетельство того, что процесс или испытания выполнены в соответствии с требованиями настоящих правил и требованиями, установленными при государственной регистрации, согласованное в письменной форме с уполномоченным лицом, отвечающим за оценку соответствия серии готовой продукции до ее выпуска;
«серия готовой продукции» (finished product batch) - в контексте настоящего документа означает серию продукции в окончательной упаковке, готовую к выпуску;
«серия нерасфасованной продукции» (bulk production batch) - серия продукции с размером, установленным при регистрации лекарственного препарата, либо готовая к фасовке в окончательную упаковку, либо находящаяся в индивидуальных упаковках и готовая для комплектования окончательных упаковок. Серия нерасфасованной продукции может содержать, например, жидкий нерасфасованный продукт, твердые лекарственные формы (таблетки или капсулы) или наполненные ампулы;
«соглашение о взаимном признании» (Mutual Recognition Agreement, MRA) - соглашение о взаимном признании инспекций со страной, в которой производятся (из которой поставляются) ввозимые лекарственные средств;
«уполномоченное лицо» (Qualified Person) - лицо, назначенное производителем лекарственных средств, которое осуществляет подтверждение соответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с требованиями Правил надлежащей практики Евразийского экономического союза. Обязанности уполномоченного лица детально описаны в главе 2 части I Правил надлежащей практики Евразийского экономического союза и в настоящем документе.
ПРИЛОЖЕНИЕ № 17
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к выпуску по параметрам
1. Принцип
1.1. Термин «выпуск по параметрам», используемый в настоящем документе, основан на определении, предложенном Европейской организацией по качеству, и подразумевает систему выпуска продукции, дающую гарантию, что продукция обладает требуемым качеством, на основании информации, полученной во время производственного процесса, а также на основании соответствия определенным требованиям Правил надлежащей производственной практики Евразийского экономического союза (далее - Правила), относящимся к выпуску по параметрам.
1.2. Выпуск по параметрам должен удовлетворять основным требованиям Правил, включая соответствующие приложения к ним и изложенные ниже требования.
2. Выпуск по параметрам
2.1. Проведение всеобъемлющего комплекса проверок и контроля параметров в процессе производства может обеспечить более высокую гарантию соответствия готовой продукции спецификации, чем испытания готовой продукции.
2.2. Выпуск по параметрам может быть разрешен в отношении некоторых специальных параметров вместо обычного испытания готовой продукции. Разрешать выпуск по параметрам, отказывать в нем или аннулировать разрешение на выпуск по параметрам должны лица, отвечающие за оценку продукции, совместно с фармацевтическим инспекторатом, ответственным за контроль соблюдения Правил.
3. Выпуск по параметрам для стерильной продукции
3.1. Настоящий раздел устанавливает требования к выпуску по параметрам готовой продукции без проведения испытания на стерильность. Непроведение испытания на стерильность является правомерным только при наличии успешного подтверждения того, что заранее установленные условия при валидации процесса стерилизации были достигнуты.
3.2. Испытание на стерильность предоставляет возможность обнаружить только значительные нарушения в системе обеспечения стерильности, что обусловлено статистическими ограничениями метода.
3.3. Выпуск по параметрам допускается только в том случае, если данные, доказывающие правильность процесса стерилизации серии, сами по себе дают достаточную гарантию того, что запланированный и валидированный процесс стерилизации обеспечивает стерильность продукции.
3.4. В настоящее время выпуск по параметрам допускается только для лекарственных препаратов, подлежащих финишной стерилизации в первичной упаковке.
3.5. Для выпуска по параметрам применяются методы стерилизации, предусматривающие в соответствии с фармакопейными требованиями использование пара, сухожарового способа и ионизирующего излучения.
3.6. Выпуск по параметрам не применяется при производстве совершенно новых лекарственных препаратов, так как удовлетворительные результаты испытаний на стерильность в течение определенного периода времени являются частью критериев приемлемости. В отдельных случаях данные испытания на стерильность, уже имеющиеся для других препаратов, можно считать достаточными для нового препарата, в который внесено только незначительное изменение с точки зрения обеспечения стерильности.
3.7. Следует выполнить анализ рисков системы обеспечения стерильности, направленный на оценку случаев выпуска нестерильной продукции.
3.8. Предыдущий опыт работы производителя лекарственных препаратов должен свидетельствовать о соответствии его производства требованиям Правил.
3.9. При оценке соответствия производства требованиям Правил следует учитывать выявленные случаи нарушения стерильности продукции, а также результаты испытаний на стерильность данного лекарственного препарата с точки зрения лекарственных препаратов, стерилизуемых таким же или аналогичным способом.
3.10. На участке производства и стерилизации, как правило, должны быть в наличии квалифицированный инженер, имеющий опыт работы по обеспечению стерильности, и квалифицированный микробиолог.
3.11. Разработка и первоначальная валидация должны гарантировать, что при соблюдении всех соответствующих условий будет выпускаться продукт одинакового качества.
3.12. Система контроля изменений должна предусматривать их рассмотрение персоналом, обеспечивающим стерильность.
3.13. Следует организовать систему контроля микробной контаминации лекарственного препарата перед стерилизацией.
3.14. Должна быть исключена возможность перепутывания продукции, прошедшей и не прошедшей стерилизацию, что обеспечивается путем физического разделения продукции или использованием электронных систем, прошедших валидацию.
3.15. Записи по стерилизации следует проверять на соответствие требованиям спецификации с использованием не менее 2 независимых систем контроля. Такой контроль может проводиться 2 сотрудниками или сотрудником и компьютерной системой, прошедшей валидацию.
3.16. Перед выпуском каждой серии продукции следует дополнительно подтвердить следующее:
все плановые работы по техническому обслуживанию и текущие проверки используемого стерилизатора выполнены;
все ремонтные работы и модификации согласованы с инженером и микробиологом, которые несут ответственность за процесс стерилизации;
используемые приборы прошли калибровку (поверку);
стерилизатор на настоящий момент валидирован для стерилизации загрузки данного типа.
3.17. Если выдано разрешение на выпуск серии продукции по параметрам, то решения о выпуске или отклонении серии продукции должны быть основаны на требованиях утвержденных спецификаций в части выпуска по параметрам. При невыполнении этих требований выпуск продукции по параметрам не допускается даже при условии успешного проведения испытания на стерильность.
Термины и определения
Для целей настоящих Требований используются основные понятия, которые означают следующее:
«выпуск по параметрам» (parametric release) - система выпуска продукции, дающая гарантию, что продукция обладает требуемым качеством, на основании информации, полученной во время производственного процесса, а также на основании соответствия определенным требованиям настоящих Правил, относящимся к выпуску по параметрам;
«система обеспечения стерильности» (sterility assurance system) - комплекс мер по обеспечению стерильности продукции. Для лекарственных препаратов, подлежащих финишной стерилизации, этот комплекс мер включает в себя следующее:
a) разработка лекарственного препарата;
b) знание и по возможности контроль микробиологических характеристик исходного сырья и технологических вспомогательных средств (например, газов и смазочных материалов),
c) проведение контроля контаминации в ходе технологического процесса для предотвращения попадания микроорганизмов в продукцию и их размножения. Это обычно достигается путем очистки и санитарной обработки поверхностей, контактирующих с продукцией, предупреждения контаминации из воздуха посредством проведения работ в чистых помещениях, проведением технологического процесса с ограничениями во времени и в соответствующих случаях использованием стадий фильтрации;
d) предотвращение перепутывания производственных потоков стерилизованной и нестерилизованной продукции;
e) постоянное достижение качества продукции;
f) процесс стерилизации;
g) система качества в целом, в том числе система обеспечения стерильности (контроль изменений, обучение персонала, наличие письменных инструкций, контроль при выпуске продукции, плановое профилактическое техническое обслуживание, анализ сбоев в работе, предотвращение ошибок по вине персонала, валидация, калибровка (поверка) и др.).
ПРИЛОЖЕНИЕ № 19
к Правилам надлежащей
производственной практики
Евразийского экономического союза
ТРЕБОВАНИЯ
к контрольным и архивным образцам
1. Область применения
1.1. Настоящий документ устанавливает требования к отбору и хранению контрольных образцов исходного сырья, упаковочных материалов или готовой продукции и архивных образцов готовой продукции.
1.2. Специальные требования к лекарственным препаратам для клинических исследований приведены в приложении № 13 к Правилам надлежащей производственной практики Евразийского экономического союза.
1.3. Настоящий документ также содержит указания в отношении отбора архивных образцов для параллельно импортируемых (поставляемых) лекарственных препаратов.
2. Принцип
2.1. Образцы хранят с целью обеспечения наличия образца для аналитических исследований и обеспечения наличия образца полностью готовой продукции. С учетом цели хранения образцы, могут быть поделены на 2 категории:
контрольный образец (reference sample) - образец, отобранный из серии исходного сырья, упаковочного материала или готовой продукции, который хранится для проведения анализа в течение срока годности серии в случае возникновения такой необходимости. Следует сохранять образцы, отобранные на критических промежуточных стадиях (например, после которых предусматриваются проведение аналитических исследований и выдача разрешений на выпуск), и промежуточных продуктов, которые поставляются за пределы зоны контроля производителя, если стабильность образцов это допускает.
архивный образец (retention sample) - образец в окончательной упаковке, отобранный из серии готовой продукции. Его хранят в целях подтверждения идентичности. Например, в течение срока хранения серии может потребоваться осмотр образца или упаковки, маркировки, инструкции по применению, получение информации о номере серии и сроке годности. Могут быть исключительные обстоятельства, когда это требование может быть соблюдено без хранения дубликатов образцов, например, если небольшие серии упаковывают для разных рынков или при производстве очень дорогих лекарственных препаратов.
Во многих случаях контрольные и архивные образцы готовой продукции идентичны и являются единицами продукции в окончательной упаковке. В таких случаях контрольные и архивные образцы могут рассматриваться как взаимозаменяемые.
2.2. У производителя, импортера или на предприятии, где выдается разрешение на выпуск серии (как указано в пунктах 7и 8настоящего документа) должны храниться контрольные и (или) архивные образцы каждой серии готовой продукции, а у производителя - контрольные образцы каждой серии исходных материалов (кроме исключений, в соответствии с пунктом 3.2настоящего документа) и (или) промежуточной продукции. На каждом предприятии, осуществляющем упаковку, следует хранить контрольные образцы каждой серии первичных упаковочных материалов и печатных материалов. Допускается включать печатные материалы в состав контрольных и (или) архивных образцов готовой продукции.
2.3. Контрольные и (или) архивные образцы выступают в качестве свидетельства о серии готовой продукции или исходных материалов и могут быть оценены в том числе в случаях, предъявления претензий в отношении качества лекарственного препарата, запросов о соответствии регистрационному досье, запросов о маркировке (упаковке) или отчета о мониторинге безопасности лекарственного препарата.
2.4. Следует вести записи для обеспечения прослеживаемой образцов; записи должны быть доступны уполномоченным органам.
3. Длительность хранения
3.1. Контрольные и архивные образцы каждой серии готовой продукции следует хранить в течение срока годности серии и 1 года после истечения срока годности. Контрольный образец должен быть упакован в его первичную упаковку или в упаковку, состоящую из того же материала, что и первичная упаковка, в которой выпускается лекарственный препарат (требования в отношении импортируемых ветеринарных лекарственных препаратов, за исключением иммунобиологических лекарственных препаратов, предусмотренных в пунктах 8 и 9 приложения № 4 к Правилам надлежащей производственной практики Евразийского экономического союза).
3.2. Образцы исходного сырья (кроме растворителей, газов или воды, предназначенных для технологических целей) должны храниться в течение не менее 2 лет после выпуска лекарственного препарата, если более длительный период не предусмотрен соответствующими нормативными правовыми актами государств-членов. Указанный период может быть сокращен, если в спецификации указан более короткий период стабильности сырья. Упаковочные материалы должны храниться в течение срока годности соответствующего готового продукта.
4. Количество контрольных и архивных образцов
4.1. Количество контрольных образцов должно быть достаточным для проведения не менее чем 2-кратного полного аналитического контроля серии продукции в соответствии с требованиями регистрационного досье, оцененного и утвержденного уполномоченным органом (организацией) государства-члена. В случае необходимости следует для каждого вида аналитического контроля использовать невскрытые упаковки. Любые исключения из этого требования должны быть обоснованы и согласованы с соответствующим уполномоченным органом (организацией) государства- члена.
4.2. Необходимо соблюдать требования в отношении количества контрольных образцов и, при необходимости, архивных образцов.
4.3. Контрольные образцы должны быть представительными для серии исходного сырья, промежуточной или готовой продукции, из которой они отобраны. Для контроля наиболее критических этапов процесса (например, начала или конца процесса) могут отбираться дополнительные образцы. Если процесс упаковки серии ведется в ходе двух и более отдельных операций по упаковке, то после каждой из этих операций следует отбирать не менее одного архивного образца. Любые исключения из этого требования должны быть обоснованы и согласованы с соответствующим уполномоченным органом государств- членов.
4.4. Необходимо обеспечить, в течение 1 года после истечения срока годности последней произведенной серии были легко доступны необходимые аналитические материалы и оборудование с целью проведения всех указанных в спецификации испытаний.
5. Условия хранения
5.1. Хранить контрольные образцы готовой продукции и активных фармацевтических субстанций следует в соответствии с требованиями законодательства государств-членов.
5.2. Условия хранения должны соответствовать требованиям, установленным при регистрации лекарственного средства (например, хранение при пониженной температуре (при необходимости).
6. Письменные соглашения
6.1. Если держатель регистрационного удостоверения не является одновременно юридическим лицом, ответственным за выпуск серии продукции в государствах-членах, обязанность по отбору и хранению контрольных (архивных) образцов должна быть определена в письменном соглашении между 2 сторонами в соответствии с главой 7 части I Правил надлежащей производственной практики Евразийского экономического союза. Указанное положение применяется также в случаях, если какая-либо деятельность по производству или выпуску серии продукции проводится не на том предприятии, которое несет ответственность за серию продукции, обращающуюся на общем рынке Союза. Порядок отбора и хранения контрольных и архивных образцов для каждого предприятия, вовлеченного в производство, должен быть определен в письменном соглашении между ними.
6.2. Уполномоченное лицо, которое выдает разрешение на выпуск серии лекарственного препарата, должно гарантировать, что все контрольные и архивные образцы будут доступны в течение приемлемого времени. Все требования в отношении такого доступа устанавливаются в письменном соглашении (при необходимости).
6.3. Если в производстве готовой продукции принимает участие более 1 производственной площадки, наличие письменных соглашений является основным требованием к отбору и месту хранения контрольных и архивных образцов.
7. Контрольные образцы. Общие положения
7.1. Контрольные образцы предназначены для проведения анализа и должны быть легко доступны для лаборатории, имеющей валидированные методики его проведения. Образцы исходного сырья, используемого в производстве лекарственных препаратов в государствах-членах, и образцы готовой продукции должны храниться на предприятии - производителе готовых лекарственных препаратов.
7.2. В отношении готовой продукции, произведенной в странах, не являющимися членами Союза:
7.2.1. Если страна имеет соглашение о взаимном признании с государствами-членами, контрольные образцы могут отбираться и храниться на предприятии-производителе. Это должно быть оформлено письменным соглашением (как указано выше в разделе 6 настоящего документа) между импортером внутри государств-членов и производителем, находящимся за пределами Союза.
7.2.2. Если страна не имеет соглашения о взаимном признании с государствами-членами Союза, то контрольные образцы готовой продукции следует отбирать и хранить на уполномоченном предприятии, расположенном в государствах-членах Союза. Отбор образцов должен выполняться в соответствии с письменным соглашением между всеми заинтересованными сторонами. Рекомендуется хранить образцы там, где проводился контроль продукции при ее ввозе.
7.2.3. Контрольные образцы исходного сырья и упаковочных материалов следует хранить там, где они использовались для производства готовых лекарственных препаратов.
8. Архивные образцы. Общие положения
8.1. Архивные образцы должны представлять серию готовых лекарственных препаратов в том виде, в котором она реализуется в государствах-членах и может понадобиться для контроля с целью подтверждения соответствия требованиям, установленным при государственной регистрации, или требованиям законодательства государств-членов (за исключением технических характеристик продукции). В связи с этим архивные образцы должны храниться на территории государств-членов. Рекомендуется хранить их в месте нахождения уполномоченного лица, выдавшего разрешение на выпуск продукции.
8.2. С учетом пункта 8.1 настоящего документа, при наличии действующего соглашения о взаимном признании и в случае, если контрольные образцы хранятся у производителя, находящегося за пределами Союза (см. пункт 7.2.2 настоящего документа), отдельные архивные образцы должны храниться в государствах-членах, если это предусмотрено законодательством этого государства-члена.
8.3. Архивные образцы должны находиться на предприятии, имеющем лицензию на производство лекарственных средств, и быть доступными для представителей уполномоченных органов.
8.4. Если при выполнении операций в последовательности «ввоз - процесс упаковки - контроль - выпуск серии» участвует более 1 производителя на территории государств-членов, ответственность за отбор и хранение архивных образцов определяется письменным соглашением (соглашениями) между участниками.
9. Контрольные и архивные образцы продукции, параллельно
импортируемой (параллельно поставляемой) дистрибьютором
9.1. Если вторичная упаковка лекарственного препарата не вскрывается, то следует хранить только используемый упаковочный материал, поскольку риск перепутывания продукции незначителен или отсутствует.
9.2. Если вторичная упаковка вскрывается (например, для замены картонной пачки или инструкции по применению), следует отбирать 1 архивный образец для каждой операции процесса упаковки, так как существует риск перепутывания продукции в процессе упаковки. Следует предусмотреть порядок, позволяющий быстро определить виновного в перепутывании (производитель или дистрибьютор), так как от этого зависит объем отзываемой продукции.
10. Контрольные и архивные образцы в случае ликвидации
предприятия-производителя
10.1. В случае ликвидации предприятия-производителя и отзыва (аннулирования, истечения срока действия) лицензии на производство в продаже может остаться большое количество серий произведенных этим производителем лекарственных препаратов с неистекшим сроком годности. Для остающихся на рынке серий производитель должен составить детальные соглашения по передаче контрольных и архивных образцов (а также соответствующей документации, касающейся настоящих Правил) на уполномоченный участок по хранению. Производитель должен согласовать с уполномоченным органом достаточность принятых мер по хранению и возможность передачи образцов для проведения оценки и анализа (при необходимости)
10.2. Если производитель не может предпринять указанные меры, то выполнение необходимых действий может быть передано другому производителю. Держатель регистрационного удостоверения несет ответственность за указанную передачу функций и представление необходимой информации уполномоченному органу (организации) государства-члена. Кроме того, держатель регистрационного удостоверения должен согласовать с уполномоченным органом государства-члена, на территории которого хранится серия с не истекшим сроком годности, достаточность мер по хранению контрольных и архивных образцов.
10.3. Эти требования распространяются также на случай ликвидации производства, находящегося за пределами государств- членов. В этом случае импортер несет особую ответственность за обеспечение того, что указанные соглашения имеют место и что проведено согласование с соответствующим(и) уполномоченным(и) органом(ами) (организациями) государств-членов.
11. Определения
Понятия, используемые в настоящем документе, означают следующее:
«баланс» (reconciliation) - соотношение между количеством продукции или материалов, произведенных или использованных теоретически и фактически, принимая во внимание обычную вариабельность;
«баллон» (cylinder) - контейнер, предназначенный для хранения газа под высоким давлением;
«банк клеток» (cell bank) - система, посредством которой производят последовательные серии продукции с использованием клеточных культур, принадлежащих одному и тому же главному банку клеток. Для приготовления рабочего банка клеток используется некоторое число контейнеров из главного банка клеток. Систему банка клеток валидируют в отношении количества пересевов или количества удвоений популяции, до достижения которых они могут использоваться в текущем производстве;
«главный банк клеток» (master cell bank) - полностью охарактеризованная культура клеток, распределенная в контейнеры за одну операцию, обрабатываемая вместе таким образом, чтобы обеспечить ее однородность, и сохраняемая таким способом, чтобы обеспечить стабильность. Обычно главный банк клеток хранят при температуре минус 70 0 С или ниже;
«рабочий банк клеток» (working cell bank) - культура клеток, отобранная из главного банка клеток и предназначенная для подготовки клеточных культур, используемых в технологическом процессе. Обычно рабочий банк клеток хранится при температуре минус 70 0 С или ниже;
«биологические агенты» (biological agents) - микроорганизмы (включая микроорганизмы, полученные методами генной инженерии), клеточные культуры и эндопаразиты, патогенные и непатогенные;
«биореактор» (biogenerator) - закрытая система, такая как ферментатор, в которую вводят биологические агенты наряду с другим сырьем таким образом, что это приводит к их размножению или к продуцированию ими других веществ путем взаимодействия с другим сырьем. Биореакторы обычно снабжены регулирующими и контролирующими приборами, а также приспособлениями для соединения, добавления и удаления веществ;
«валидация» (validation) - действия, которые в соответствии с настоящими Правилам доказывают, что определенная методика, процесс, оборудование, сырье, деятельность или система действительно приводят к ожидаемым результатам (см. также термин «квалификация»);
«возврат» (return) - отправка назад производителю или дистрибьютору лекарственного препарата независимо от наличия дефекта;
«воздушный шлюз» (air-lock) - ограниченное пространство с двумя или несколькими дверями, расположенное между двумя или несколькими помещениями, например, разных классов чистоты, служащее для контроля потока воздуха между этими помещениями, когда в них необходимо войти. Воздушные шлюзы предназначаются и используются как для перехода персонала, так и для перемещения материалов;
«вспомогательное вещество» - вещество, за исключением активных фармацевтических субстанций, входящее в состав лекарственного препарата для придания ему необходимых свойств;
«готовая продукция» (готовый продукт) (finished product) - лекарственный препарат, который прошел все стадии технологического процесса, включая укладку в окончательную упаковку;
«запись» (record) используется в том же значении, что и в части I настоящих Правил;
«изолированная зона» (contained area) - зона, построенная и эксплуатируемая таким образом (и оборудованная соответствующими системами обработки и фильтрации воздуха), чтобы предотвратить контаминацию производственной среды биологическими агентами изнутри зоны;
«изоляция» (containment) - действия по ограничению распространения биологических агентов или других контаминантов за пределы определенного пространства;
«первичная изоляция» (primary containment) - система изоляции, которая предотвращает проникновение биологического агента в близлежащую производственную зону. Это достигается использованием закрытых контейнеров или боксов для безопасного ведения биологических работ наряду с процедурами безопасного ведения процесса;
«вторичная изоляция» (secondary containment) - система изоляции, предотвращающая проникновение биологического агента во внешнюю окружающую среду или в другие рабочие зоны. Это достигается использованием помещений со специальными системами подготовки воздуха, наличием воздушных шлюзов и (или) стерилизаторов для передачи материалов наружу наряду с процедурами безопасного ведения процесса. Во многих случаях используется для повышения эффективности первичной изоляции;
«инфицированный» (infected) - зараженный посторонними биологическими агентами и, следовательно, способный к распространению инфекции;
«исходные материалы» (starting material) - любое вещество, используемое при производстве лекарственного препарата, за исключением упаковочных материалов;
«калибровка» (calibration) - совокупность операций, проводимых при заданных условиях, посредством которых устанавливается соотношение между значениями величин, полученными с помощью средства измерений (или измерительной системой), или значениями величин, представленными материальной мерой, и соответствующими известными значениями опорного эталона (стандартного образца);
«карантин» (quarantine) - статус исходного сырья, упаковочных материалов, промежуточной, нерасфасованной или готовой продукции, изолированных физически или другими эффективными способами, до вынесения решения об их выпуске или отклонении;
«квалификация» (qualification) - действия, удостоверяющие, что конкретное оборудование работает правильно и действительно ведет к ожидаемым результатам. Понятие «валидация» является более широким и иногда включает в себя понятие «квалификация»;
«клеточная культура» (cell culture) - клеточная масса, полученная в результате выращивания in vitro клеток, изолированных от многоклеточных организмов;
«коллектор» (manifold) - устройство или оборудование, позволяющее одновременно наполнять газом несколько баллонов (контейнеров) из одного источника;
«компьютеризированная система» (computerized system) - система, включающая ввод данных, их электронную обработку и выдачу информации, используемая либо для документального оформления, либо для автоматического управления;
«контролируемая зона» (controlled area) - зона, построенная и эксплуатируемая таким образом, чтобы контролировать возможную контаминацию и случайное распространение живых организмов (может использоваться система воздухоподготовки, приблизительно соответствующая классу D). Степень осуществляемого контроля должна зависеть от природы организма, используемого в процессе. Как минимум, контролируемая зона должна эксплуатироваться при отрицательном давлении по отношению к смежным помещениям и позволять эффективно устранять незначительные количества находящихся в воздухе источников контаминации;
«контроль в процессе производства» (in-process control) - проверки, осуществляемые во время технологического процесса в целях его контроля и при необходимости регулирования для обеспечения соответствия продукции спецификациям. Контроль производственной среды или оборудования также может рассматриваться как часть контроля в процессе производства;
«контроль качества» (quality control) используется в том же значении, что и в пункте 1.4 главы 1 части I настоящих Правил;
«криогенный сосуд» (cryogenic vessel) - контейнер, предназначенный для хранения сжиженного газа при сверхнизких температурах;
«лекарственное растительное сырье» (medicinal plant) - свежие или высушенные растения, водоросли, грибы или лишайники либо их части, цельные или измельченные, используемые для производства лекарственных средств;
«лекарственное средство» (medicinal product) - средство, представляющее собой или содержащее вещество или комбинацию веществ, вступающее в контакт с организмом человека, предназначенное для лечения, профилактики заболеваний человека или восстановления, коррекции или изменения его физиологических функций посредством фармакологического, иммунологического либо метаболического воздействия или для диагностики заболеваний и состояний человека;
«лекарственный растительный препарат» (herbal medicinal product) - лекарственный препарат, содержащий в качестве активных компонентов исключительно лекарственное растительное сырье и (или) препараты на его основе;
«мониторинг производственной среды» - наблюдение за состоянием объектов производственной среды (помещения, оборудование, воздух рабочей зоны, технологические среды, персонал) для определения и (или) предсказания момента перехода в предельное состояние на основе сравнения измеренных параметров с заданными значениями;
«нерасфасованная продукция» (bulk product) - любая продукция, прошедшая все стадии технологического процесса за исключением окончательной упаковки;
«номер серии», «номер партии» (batch number or lot number) - отличительная комбинация цифр, букв и (или) символов, которая идентифицирует серию (партию) и на основании которых можно проследить историю ее производства и реализации;
«перекрестная контаминация» (cross contamination) - загрязнение исходного сырья или продукции другим исходным сырьем или другой продукцией;
«переработка» (reprocessing) - переработка всей или части серии продукции неприемлемого качества на определенной стадии технологического процесса так, чтобы ее качество могло стать приемлемым, посредством одной или нескольких дополнительных операций;
«повторное использование» (recovery) - введение произведенной ранее серии продукции требуемого качества (или ее части) в другую серию продукции на определенной стадии производства;
«посевная культура» (seed lot) - система посевной культуры (seed lot system): Система, в соответствии с которой последовательные серии продукции производят из одной и той же главной посевной культуры при определенном количестве пересевов (пассажей). Для обычного производства рабочую посевную культуру готовят из главной посевной культуры. Исходя из требований безопасности и эффективности, при производстве готовой продукции, которую получают из рабочей посевной культуры, не должно использоваться большее количество пассажей из главной посевной культуры, чем для вакцины, прошедшей клинические исследования. Происхождение и количество пассажей главной посевной культуры и рабочей посевной культуры должны быть оформлены документально;
«главная посевная культура» (master seed lot) - культура микроорганизмов, распределенная из одного объема посевной культуры в емкости в процессе одной операции таким образом, чтобы обеспечить однородность, предотвратить контаминацию и гарантировать стабильность. Главную посевную культуру в жидком виде обычно хранят при температуре минус 70 0 C или ниже, в лиофилизированном виде - при известной температуре, обеспечивающей стабильность;
«рабочая посевная культура» (working seed lot) - культура микроорганизмов, полученная из главной посевной культуры и предназначенная для использования в производстве. Рабочую посевную культуру распределяют в емкости и хранят, как описано выше для главных посевных культур;
«производитель» (manufacturer) - организация, осуществляющая деятельность по производству лекарственных средств;
«производство» (manufacture) - все операции по закупке исходного сырья, материалов и продукции, технологическому процессу, контролю качества, выдаче разрешения на выпуск, хранению, реализации лекарственных средств и по соответствующему контролю;
«производственная площадка» - территориально обособленный комплекс производителя лекарственных средств, предназначенный для выполнения всего процесса производства лекарственных средств или его определенной стадии;
«промежуточная продукция» (intermediate product) - частично обработанное исходное сырье, которое должно пройти последующие стадии производства прежде, чем оно станет нерасфасованной продукцией;
«процедура» (procedures) - описание обязательных для выполнения операций и мер предосторожности, а также всех необходимых мероприятий, осуществление которых прямо или косвенно связано с производством лекарственного средства;
«радиофармацевтическое лекарственное средство» (radio pharmaceutical) - любое лекарственное средство, которое в готовом для применения виде содержит один или несколько радионуклидов (радиоактивных изотопов), введенных в него в медицинских целях;
«серия», «партия» (batch, lot) - определенное количество исходного сырья, упаковочных материалов или продукции, подвергаемое обработке в одном или в ряде последовательных технологических процессов таким образом, чтобы рассчитывать на однородность продукции. Для завершения некоторых этапов производства иногда необходимо разделить серию на определенное количество подсерий, которые позже объединяют для получения окончательной однородной серии. При непрерывном производстве понятие серии должно относиться к определенной части продукции, характеризуемой однородностью.
Применимо также следующее определение серии в отношении контроля готовой продукции: «При контроле готовой продукции считается, что к серии лекарственного препарата относятся все единицы данной лекарственной формы, которые произведены из одного исходного количества материала и прошли одну и ту же серию производственных операций или операцию по стерилизации, или при непрерывном технологическом процессе все единицы, произведенные в данный период времени»;
«сжиженные газы» (liquefiable gases) - газы, которые при стандартных температуре и давлении наполнения находятся в баллоне в сжиженном виде;
«система» (system) - регулируемая модель взаимосвязанных действий и технических средств, объединенных для создания организованного целого;
«спецификация» (specification) используется в том же значении, что и в части I Правил надлежащей производственной практики Евразийского экономического союза;
«стерильность» (sterility) - отсутствие живых организмов. Требования к проведению контроля стерильности приведены в соответствующей фармакопее;
«тара» (container) - изделие - элемент упаковки, предназначенное для размещения продукции;
«технологический процесс» (production) - все операции, связанные с производством лекарственного средства, начинающиеся с приемки исходного сырья, продолжающиеся обработкой и упаковкой и завершающиеся получением готовой продукции;
«упаковка» (packaging) - все операции, включая фасовку и маркировку, которые необходимо пройти нерасфасованной продукции, чтобы стать готовой продукцией. Наполнение стерильной продукции, как правило, не следует рассматривать как часть процесса упаковки, поскольку в первичные упаковки продукция дозируется, но окончательно не упаковывается;
«упаковочный материал» (packaging material) - любой материал, используемый при упаковке лекарственного средства, кроме любой транспортной тары для транспортирования или отгрузки. Упаковочные материалы относятся к первичным или вторичным в зависимости от того, предназначены они для непосредственного контакта с лекарственным средством или нет;
«чистая зона» (clean area) - зона, в которой контролируется производственная среда на наличие контаминирующих частиц и микроорганизмов, построенная и эксплуатируемая таким образом, чтобы уменьшить проникновение, образование и сохранение контаминантов внутри зоны. Различные уровни контроля производственной среды установлены в соответствии с приложением № 1 к Правилам надлежащей производственной практики Евразийского экономического союза;
«чистая (изолированная) зона» (clean contained area) - зона, построенная и эксплуатируемая таким образом, что она одновременно является чистой и изолированной зоной;
«экзотический организм» (exotic organism) - биологический агент, вызывающий заболевание, отсутствующее в данной стране или географической зоне, либо являющийся объектом профилактических мер или программы по его устранению в данном государстве или в этой географической зоне.

Адрес редакции:
г. Алматы, ул. Жандосова, 98, БЦ "Навои Тауэрс",
6 этаж, офис 603
тел.:+7 727 385 85 69,
+7 708 312 70 75,
+7 708 605 47 73,
+7 747 409 77 56
Система оплаты: 

{kind=link}